在單細胞RNA測序中DoubletFinder雙細胞分析方法詳解
在單細胞RNA測序(scRNA-seq)中,當兩個細胞被捕獲到同一反應液滴時,會形成雙聯體(doublet)。在后續分析中會因帶有相同的Barcode,會被認為是一個細胞的偽細胞。這類細胞會對分析產生影響,從而扭曲分析結果。因此,計算doublet檢測方法成為必要工具。
《Benchmarking Computational Doublet-Detection Methods for Single-Cell RNA Sequencing Data》文獻系統性評估了九種主流方法(包括doubletCells、Scrublet、cxds、bcds、hybrid、Solo、DoubletDetection、DoubletFinder和DoubletDecon),使用16個真實數據集,并通過scDesign生成的112個合成數據集,從檢測的準確性,下游分析的影響,到計算效率等各方面評估。
綜合顯示,DoubletFinder在檢測準確性上表現最佳。
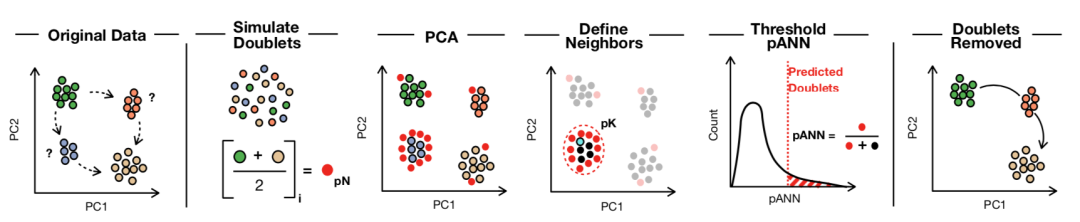
DoubletFinder分析可分為4個步驟:
(1) 利用已有的scRNA-seq數據生成doublet;
(2) 對合并的真實人工數據進行預處理;
(3) 進行PCA主成分分析,利用PC距離矩陣求出每個細胞的人工k個最近鄰(pANN)的比例;
(4) 根據預期的doublet數量排序和計算閾值pANN值。
以下是我們分析的示例代碼:
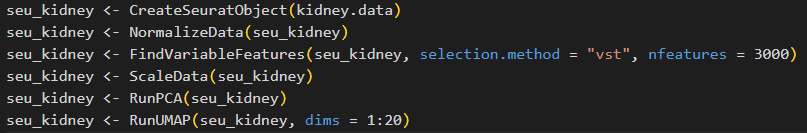
1.讀入單細胞數據,并進行一系列預處理(包括單細胞數據質控、標準化、降維聚類等):
seu_kidney <- CreateSeuratObject(kidney.data)
seu_kidney <- NormalizeData(seu_kidney)
seu_kidney <- FindVariableFeatures(seu_kidney, selection.method = "vst", nfeatures = 3000)
seu_kidney <- ScaleData(seu_kidney)
seu_kidney <- RunPCA(seu_kidney)
seu_kidney <- RunUMAP(seu_kidney, dims = 1:20)
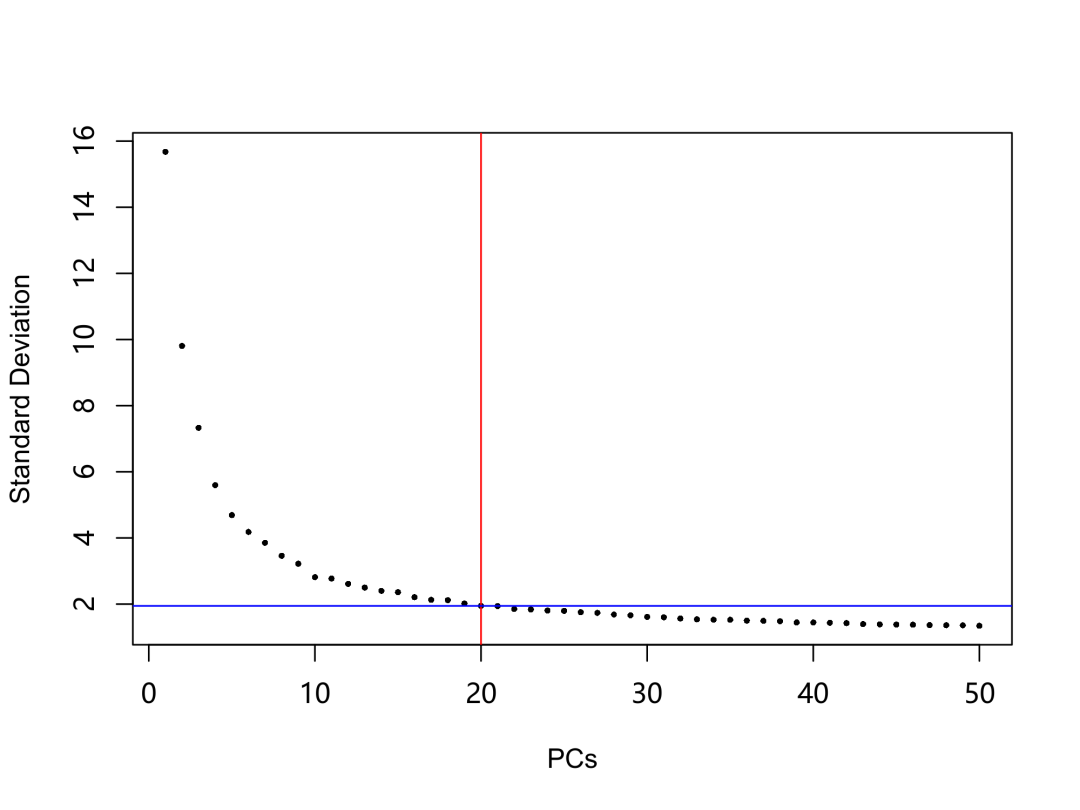
選擇統計上顯著的主成分的數量,下圖為我們選擇的主成分數量。
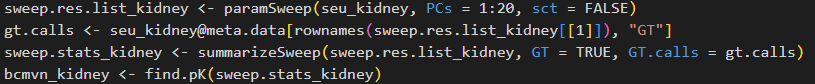
2. 尋找最優pK值:
sweep.res.list_kidney <- paramSweep(seu_kidney, PCs = 1:20, sct = FALSE)
gt.calls <- seu_kidney@meta.data[rownames (sweep.res.list_kidney[[1]]), "GT"]
sweep.stats_kidney <- summarizeSweep(sweep.res.list_kidney, GT = TRUE, GT.calls = gt.calls)
bcmvn_kidney <- find.pK(sweep.stats_kidney)
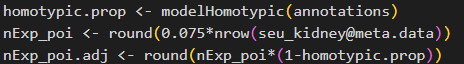
3. 雙細胞比例計算:
homotypic.prop <- modelHomotypic(annotations)
nExp_poi <- round(0.075*nrow(seu_kidney@meta.data))
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
4. 鑒定雙細胞:
seu_kidney <- doubletFinder(seu_kidney, PCs = 1:20, pN = 0.25, pK = 0.09, nExp = nExp_poi, reuse.pANN = FALSE, sct = FALSE)
seu_kidney <- doubletFinder(seu_kidney, PCs = 1:20, pN = 0.25, pK = 0.09, nExp = nExp_poi.adj, reuse.pANN = "pANN_0.25_0.09_913", sct = FALSE)

5. 確定了Doublet-Low Confidience, Doublet-High Confidience,Singlet三種細胞類型,并進行可視化,包含TSNE和UMAP兩種展示形式:
DimPlot(pbmc, reduction = "tsne", group.by ="DF_hi.lo",cols =c("red","gold","#1bb3b6"),pt.size = 0.8) + ggtitle("DoubletFinder")
DimPlot(pbmc, reduction = "umap", group.by ="DF_hi.lo",cols =c("red","gold","#1bb3b6"),pt.size = 0.8) + ggtitle("DoubletFinder")

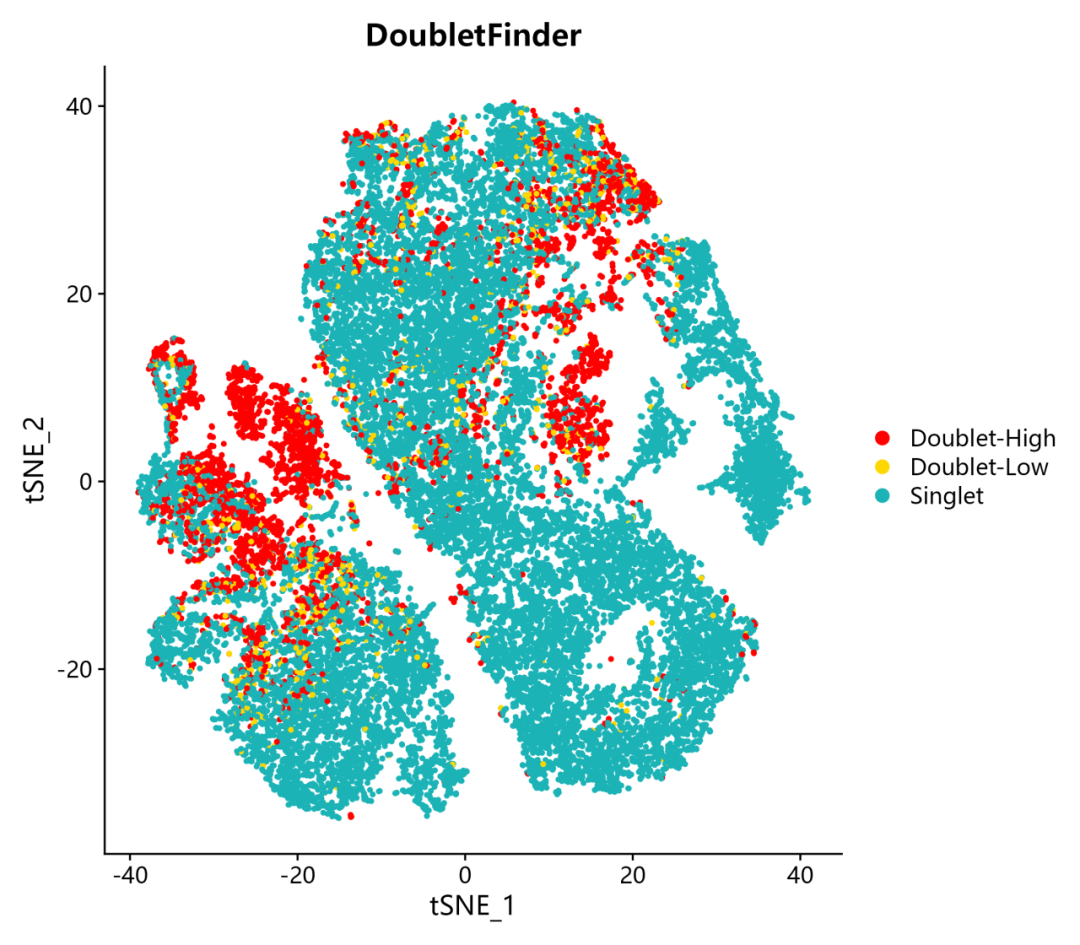
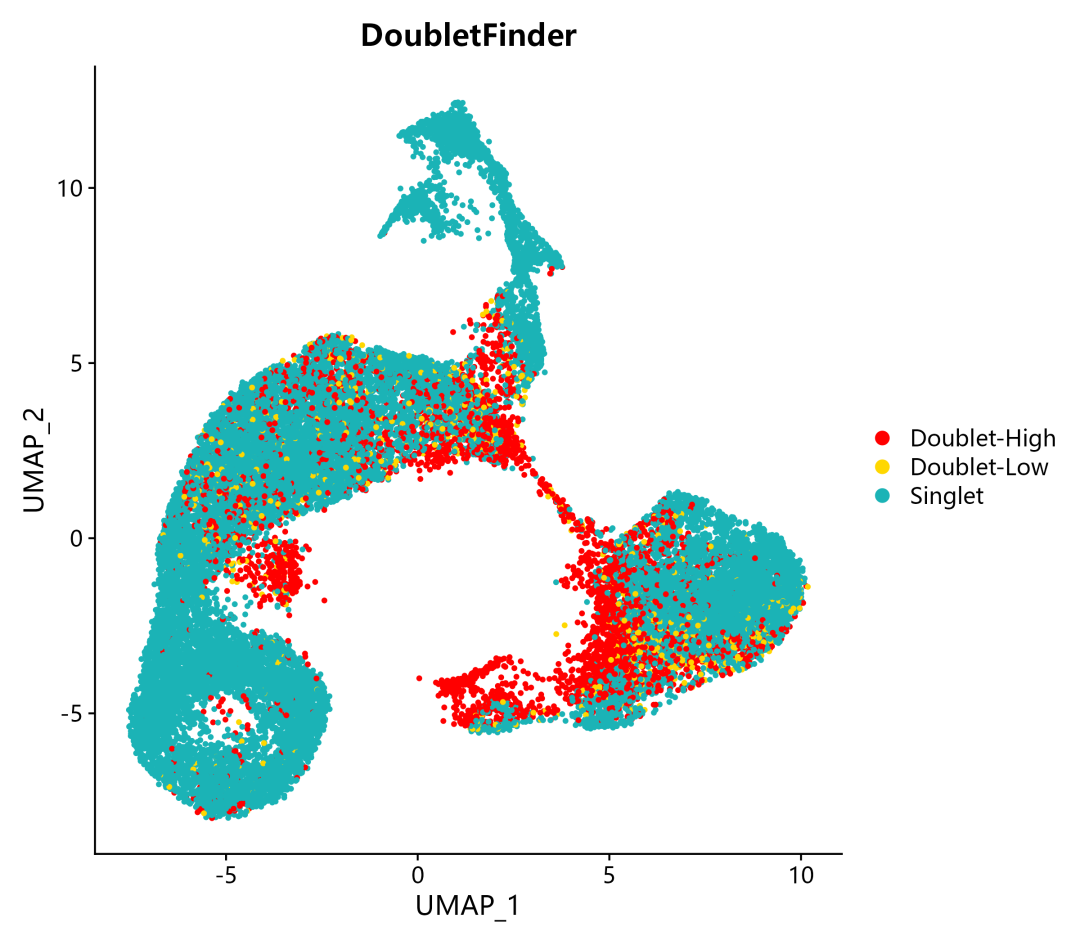
在完成DoubletFinder雙細胞分析之后,我們可以從單細胞數據的meta.data表中,查看結果,用于之后的下游分析。
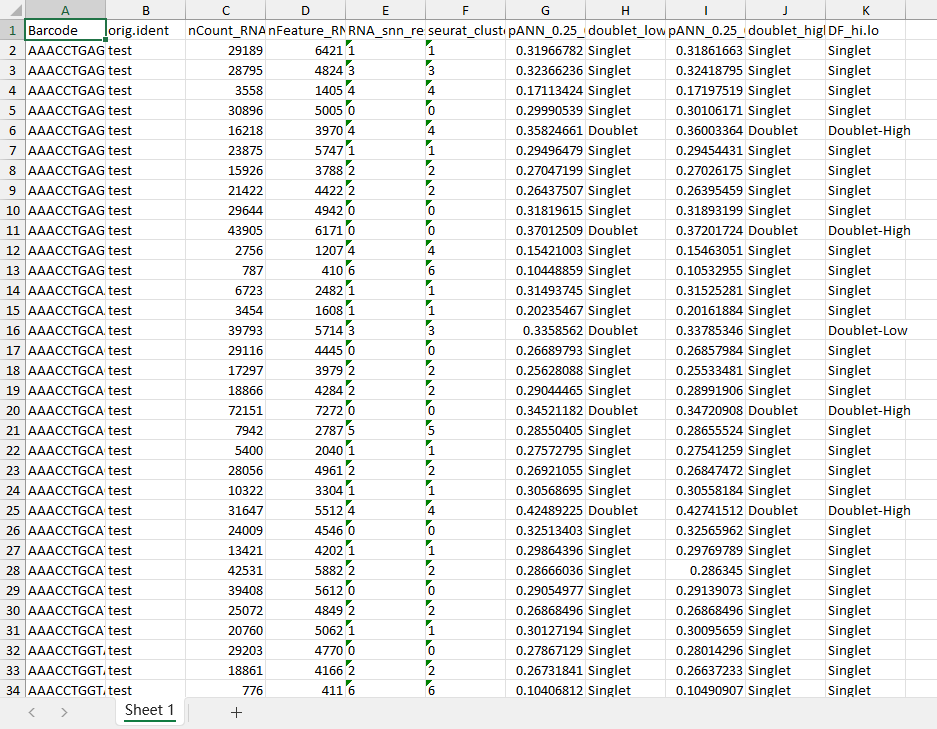
以上是對DoubletFinder雙細胞分析方法的展示。雙細胞的去除非常必要,當我們剔除這類潛在的雙細胞數據后,后續的分析結果也就更可靠了。