時空組學研究進展(七):單細胞空間轉錄組學技術的介紹

期刊:Science China-Life Sciences
影響因子:8.0
在前面的章節中,我們系統地回顧了單細胞組學的最新進展。盡管這些單細胞測序技術允許以前所未有的分辨率調查細胞異質性,但它們遠遠不足以充分了解多細胞生物的復雜工作原理。許多研究強調,一個細胞的狀態不僅受到細胞內調節網絡的調節,還受到來自環境的細胞外信號的干擾。在實驗過程中,組織的分離和單個細胞的分離都會導致關鍵空間信息的丟失,包括細胞位置及其相互接近度。空間轉錄組學(ST)解決了這一限制,使測量基因表達與空間信息保存。在本節中,我們將介紹空間轉錄組學技術,討論空間數據分析的計算方法,并回顧其在各種生物系統中的應用。此外,我們還將深入探討空間多組學技術的最新進展。
空間分辨轉錄組學技術
目前所有的空間轉錄組學技術可以大致概括為三類,主要基于(i)顯微解剖,(ii)條形碼,和(iii)成像(圖14A-C)。這些ST技術在位置標記和轉錄本分析的方法上有所不同,這可能決定了空間分辨率、檢測效率、要求的樣本類型等等。接下來,我們將討論從每個類別中選擇代表性技術的原則,并總結它們的特點。整理的技術列表及其相應的特征如表4所示。
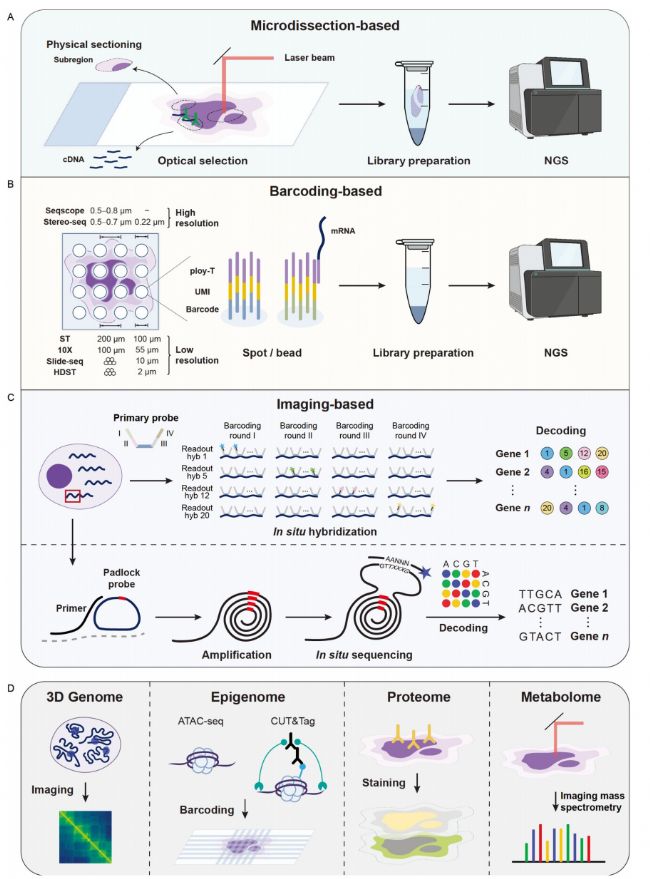
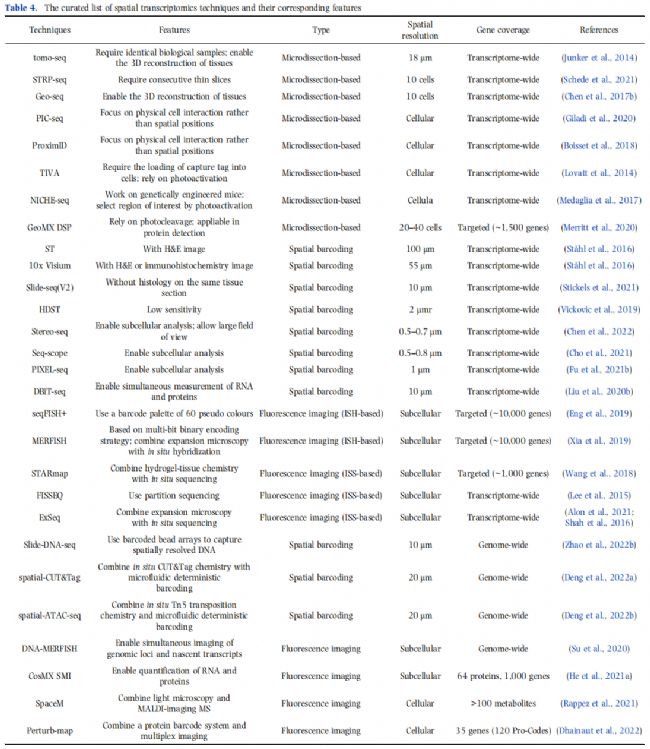
1. 基于顯微解剖的ST技術
屬于這一類的技術旨在通過各種顯微解剖方法從多個空間近端組織亞區中計算重建組織的三維結構(圖14A)。例如,RNA斷層掃描(tomoseq)從多個假定相同的生物樣本中沿著三個正交軸的一系列順序冷凍中獲得RNA。相同生物樣本的要求限制了tomo-seq在人類樣本上的應用。相比之下,STRP-seq采用兩級解剖策略將組織切片分為初級切片和次級切片,假設空間表達模式在間隔14 μm的連續初級切片之間是恒定的。基于冷凍切片,Geo-seq利用LCM將組織切片成小至10個細胞左右的區域。這類方法中的其他方法包括ProximID 和PIC-seq,它們側重于兩個(雙胞胎)或三個細胞(三胞胎)內的物理細胞相互作用,而不是組織中的位置或周圍環境。
除了物理切片,顯微解剖可以通過結合光學標記和基于熒光的細胞選擇,或基因指數寡核苷酸的光切割來完成。例如,轉錄組體內分析(TIVA)加載TIVA標簽(即光激活的mRNA捕獲分子)進入活細胞,并通過激光光激活選擇細胞,隨后觸發標簽與mRNA的雜交。作為一種替代技術,NICHE-seq將標記的地標細胞注射到表達光激活綠色熒光(PA-GFP)的轉基因小鼠中,允許對感興趣的生態位進行原位標記。組織解離后,活化的PA-GFP+細胞通過FACS進行分類,進行單細胞轉錄組分析。NanoString開發的商用GeoMX數字空間輪廓儀(DSP)采用帶有UV可切割接頭的探針,并自動進行光學選擇。
總的來說,微解剖與單細胞或bulk-RNA相結合測序使得在空間背景下研究轉錄組成為可能。顯微解剖可以用物理方式進行,也可以用光學方式進行。物理切片通常是手工進行的,這使得解剖方案既費力又耗時。相比之下,光學依賴切片通常依賴于將專門的標簽加載到活細胞或模式生物的基因工程中,這限制了其在新鮮冷凍或FFPE人類樣本中的應用。無論如何進行顯微解剖和測序,在所選擇的子區域內的剖面細胞的確切位置是未知的,導致普遍較低的空間分辨率。
2. 基于條形碼的ST技術
基于顯微解剖的技術通過手動標記每個子區域來跟蹤空間信息。空間條形碼技術可以自動記錄空間坐標(圖14B)。在這種方法中,條形碼與UMIs和聚寡核苷酸一起固定在玻璃載玻片上,以便原位捕獲mRNA和cDNA合成。陣列中的每個條形碼點直徑為100 μm,距離相鄰點的中心距離為200 μm,分辨率為10-40個單元。10x Genomics利用直徑為55 μm、中心到中心距離為100 μm的斑點進一步將空間分辨率提高到5-10個細胞。一些技術不是將條形碼直接附著在載玻片上,而是將條形碼與小珠連接起來,用于位置標記和mRNA捕獲。例如,Slide-seq將10 μm的dna條形碼珠沉積在表面上。同樣,HDST將條形碼珠放入2 μm井的陣列中。這兩種技術都將空間分辨率提高到1-2個單元。然而,由于條形碼珠粒是隨機分布在載玻片上的,因此需要原位測序(ISS)或原位雜交(ISH)來解碼每個固定珠粒的條形碼序列。盡管基于頭部的技術可以達到細胞分辨率,但它們仍然過于粗糙,無法檢測亞細胞差異。
最近,通過重新利用Illumina測序平臺,開發了Seq-scope,以實現0.5-0.8 μm的中心到中心分辨率。另一種實現亞微米分辨率分析的技術是Stereo-seq,其中包含條形碼的220納米DNA納米球(dnb)沉積在中心距離為500或715納米的圖案陣列上。Seq-scope和Stereo-seq都需要兩輪測序,其中第一輪將條形碼與空間位置相關聯,第二輪提供捕獲cDNA的信息,就像Slide-seq一樣。
總之,基于條形碼的方法將空間條形碼技術與NGS相結合,允許在空間背景下對RNA進行轉錄組分析。這些技術涉及空間分辨率和檢測效率之間的權衡。與最初的ST技術或商業化的10倍Visium相比,Seq-scope、Stereo-seq在空間分辨率上的提高往往是以低檢測靈敏度和低基因覆蓋率為代價的。
3. 基于成像的ST技術
基于微解剖和基于條形碼的技術在位置標記后提取核酸分子用于NGS測序。為了在原位保存RNA,各種原位轉錄組學技術被開發出來用于基因表達的空間定位,包括ISH和ISS(圖14C)。由于這些方法需要熒光成像,因此它們被統稱為基于成像的技術。大多數基于ish的ST技術主要依靠單分子RNA熒光原位雜交(smFISH)來實現靶向轉錄物的原位定量測量。SeqFISH屬于這種類型,它可以通過連續的熒光雜交、成像和剝離讀出探針來同時檢測多個mRNA分子。使用seqFISH策略,所有的目標基因都是通過幾輪讀出探針的組合來編碼的。SeqFISH+將讀出探針調色板從SeqFISH中的四種或五種顏色擴展到60種“偽顏色”,從而在單個細胞中實現多達10000個基因的多路復用。MERFISH是另一種基于smfish的技術,它也需要多輪雜交,但采用了獨特的多位二進制編碼策略。為了解決光學擁擠問題,將擴展顯微鏡(ExM)集成到MERFISH中。編碼策略,結合ExM,允許MERFISH減少雜交輪數。例如,為了保證檢測到10,000個基因,使用三色成像,seqFISH+需要80輪(4×20)雜交,而MERFISH只需要23輪就能構建一個漢明權值為4的69位HD4編碼。除了基于多路FISH的技術外,ISS也可以實現RNA的原位分析,它通過原位信號擴增對固定組織或細胞樣本中的RNA進行測序。由于細胞空間有限,一些基于isss的技術通過設計針對特定RNA或cDNA的探針來選擇部分基因。2013年發表的最初的ISS方法使用掛鎖探針與靶標結合,然后通過滾環擴增(RCA)產生RCA產物,用于后續的結扎測序。STARmap使用雙組分掛鎖探針直接結合RNA而不是cDNA,避免了RNA到cDNA的低效步驟,降低了潛在的噪聲。為了消除傳統的支持寡核苷酸連接檢測(SOLiD)測序帶來的強背景熒光,STARmap設計了動態退火和連接減錯測序(SEDAL),可以在測序過程中抑制誤差。
除了靶向ISS方法外,還可以采用非靶向方式進行ISS,即將轉錄物反向轉錄為cDNA,然后進行DNA擴增和測序,而不需要對基因進行預選擇。雖然非靶向方式可以提高轉錄組的覆蓋范圍,但它也可能導致分子擁擠。為了緩解這一問題,FISSEQ利用了分區測序策略,其中只有一小部分擴增子被隨機選擇并使用擴展測序引物進行測序,因此導致檢測效率較低。結合ExM, FISSEQ適用于另一種稱為ExSeq的方法,以區分擁擠的分子并提高空間分辨率。
一般來說,基于成像的技術提供高空間分辨率,達到細胞甚至亞細胞水平。在這些技術中,基于ish的技術依賴于目標基因的先驗知識,具有較高的檢測效率。相比之下,由于國際空間站的局限性,基于國際空間站的技術已經效率相對較低,特別是在沒有目標的情況下。此外,大多數這些技術都需要專門的高分辨率成像設備,這可能限制了它們更廣泛的適用性。
4. 空間多組學技術
為了對細胞進行更全面的表征,在空間背景下對其他模式的測量已經付出了相當大的努力,包括基因組、表觀基因組、蛋白質組、代謝組等(圖14D)。ST技術中使用的定位策略已經適應于實現其他組學的空間分析。例如,SlideDNA-seq使用條形碼頭陣列捕獲空間分辨的基因組序列,該陣列最初是為空間RNA分析而開發的。同樣,通過將DbiT-seq的微流體確定性條形碼策略與原位CUT&Tag化學和Tn5轉位化學相結合,開發了spatial-CUT&Tag 和spatial-ATAC-seq 來分析組蛋白修飾和染色質可及性。為了了解其原生環境下的三維染色質構象,設計了一種基于merfish的方法來可視化超過1000個基因組位點,用于高分辨率染色質追蹤。
在蛋白質組學領域,蛋白質表達可以很容易地通過多重免疫組織化學(IHC)可視化。免疫組化可以進一步與成像質細胞術或多路離子束成像(MIBI)相結合,允許同時成像約100種蛋白質。此外,感興趣的蛋白質可以被dna條形碼抗體靶向,從而通過NGS進行量化,如GeoMx DSP 。細胞表面蛋白可以被抗體結合而不產生細胞裂解,從而防止RNA受到損傷。因此,無論是單細胞組學還是空間組學,蛋白質組學都可以與轉錄組學相結合。例如,增強版的10x Visium在mRNA捕獲之前進行免疫組化,以實現蛋白質和RNA的共同檢測,盡管只允許檢測1-2種蛋白質。通過在流動條形碼之前將抗體衍生標簽添加到固定組織載片,DbiT-seq可以測量mRNA和數十種蛋白質。此外,NanoString還提供CosMx SMI平臺,可通過高plex成像對1000種RNA和64種蛋白質進行定量分析。從樣品中收集的代謝物通常使用質譜法進行定量。為了研究空間分辨代謝組,基于成像質譜法(IMS)的各種技術已經發展起來。這些技術在從樣品分子中產生離子的方式上有所不同,包括MALDI 、DESI和SIMS。例如,SpaceM是一種基于maldi的原位單細胞代謝組學方法。它通過將maldi成像與光學顯微鏡相結合,然后使用計算方法進行圖像分割和配準,解決了將代謝物強度分配給單個細胞的挑戰。
除了內在遺傳外,許多基因功能還受到空間環境的影響。為了研究空間功能基因組學,Dhainaut建立了一種名為Perturb-map的方法,該方法可以在組織背景下以單細胞分辨率匯集CRISPR篩選。這是通過采用蛋白質條形碼系統和多路成像來實現的。
空間轉錄組學計算方法
單細胞分析的標準工作流程包括關鍵任務,如細胞聚類、細胞類型注釋、差異表達分析、譜系追蹤、細胞-細胞通信和集成分析。這些任務也構成了ST數據分析的主干。空間轉錄組學以其獨特的能力提供空間接近性和環境信息,不僅拓寬了分析范圍,也帶來了巨大的整合挑戰。為了解決這些問題,已經開發了大量的計算方法來整合基因表達與空間信息,并提供新的見解(圖15)。我們將在接下來的章節中回顧為不同目的而設計的方法。已發表的計算方法列表載于支持資料表S11。
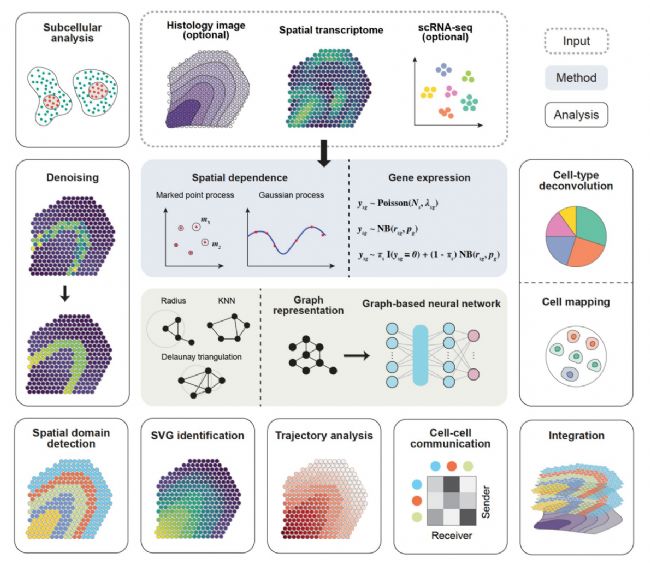
1. 去噪增強空間轉錄組學中的信號
如上所述,許多ST技術面臨著與低檢測效率和顯著噪聲相關的挑戰。這些問題源于對每個空間單元(即點或頭)的淺層測序或保存組織結構所需的復雜實驗步驟,或兩者的結合。Wang等人在10倍的Visium和Slide-seq數據中表明,信號噪聲反映在基因表達的dropouts和膨脹中。雖然已經為scRNA-seq數據開發了去噪方法來解決drop-out問題,但它們往往難以糾正“假的”高表達。此外,這些單細胞方法僅依賴于轉錄組學數據,因此不能直接應用于整合額外的空間信息。
專門開發了幾種計算方法來處理去噪的ST數據。例如,spprod可以通過基于潛在圖學習的基于條形碼的技術在噪聲ST數據中推算基因表達。spprod中的去噪過程包括兩個步驟。首先,spprod通過利用空間接近性和表達相似性來構建一個圖。重要的是,如果可以的話,從相應的病理圖像中提取的特征可以用于圖的構建。接下來,spprod通過借用圖中顯示的鄰域的表達信息來糾正每個點/頭的基因表達。另一種方法,spARC,采用了類似的基于圖形的框架,但證明了其在基于成像的ST技術上的適用性。SiGra也是一種基于圖的方法,但采用了不同的方法來構建圖。它利用成像、轉錄組和混合三個圖形轉換器自編碼器以及注意機制,使SiGra能夠用多模態空間信息增強稀疏和嘈雜的轉錄組數據。stLearn中的SME方法還允許整合圖像特征來規范化空間基因表達。它采用了一種簡單的加權平均策略,根據接近點之間的形態相似性計算權重。Ni等人認為,損失和膨脹不是隨機的輟學或膨脹,而是由附近點之間和點之間的mRNA流血造成的,這被稱為點交換。為了調整現貨交換的影響,他們提出了一種稱為SpotClean的方法。SpotClean采用概率框架來模擬給定位點上的基因特異性表達,該框架考慮了該位點組織中存在的reads,并將其讀取到其他位點上的出血,同時也將其他位點上的出血去除。作者證明SpotClean可以在ST和10x Visium等技術中準確估計基因特異性UMI計數,其中背景位置可以通過ST幻燈片與匹配的H&E圖像之間的比對來識別。
2. 基于成像的ST數據的亞細胞分析
基于成像的ST技術為細胞甚至亞細胞分析提供了很好的機會,但也帶來了很大的挑戰。對于這些技術,每個測量的像素只代表一個轉錄本,這不足以推斷它屬于細胞類型。如何將這些單個像素合并形成細胞或亞細胞結構將具有重要意義。在目前的研究中,有兩種主要策略用于高分辨率ST數據的分析:基于分割的方法或無分割的方法。細胞分割最初是在顯微鏡免疫組化圖像處理中提出的,它提供了更多關于細胞數量和細胞形態的信息。這里的細胞分割是基于轉錄本的稀疏測量來確定細胞邊界,即將轉錄本分配給細胞。傳統的細胞分割依賴于從染色圖像中提取的特征,包括強度和紋理,其中一些可以代表細胞邊界。但是對于RNA的熒光圖像,揭示細胞邊界需要對細胞膜進行特定的染色,這阻礙了細胞的分割。大多數組選擇進行額外的細胞核染色(例如DAPI)來識別假定的細胞核,然后用于指導轉錄本分配。考慮到基因在細胞核區域的表達可能不等于在整個細胞內的表達,一些組合并,輔助poly(A)染色以告知細胞的體細胞。已經開發了幾種計算方法來提供替代解決方案。
例如,Qian等人開發了pciSeq,它利用概率框架將RNA點分配給其原始細胞。具體而言,pciSeq將DAPI圖像中的細胞核分割作為細胞的初始近似,并分別通過負二項分布和泊松過程模擬細胞RNA計數和基因-細胞距離。該方法以配對的scRNA-seq為參考,利用變分貝葉斯推理估計轉錄本同時屬于細胞和細胞類型的概率。JSTA是另一種依賴于DAPI染色的初始細胞核分割和匹配的scRNA-seq參考的方法。JSTA還可以利用深度神經網絡(deep neural network, DNN)作為分類器,通過迭代像素分配實現聯合細胞分割和細胞類型標注。
細胞分割可以以不依賴于scrna的方式實現。例如,Baysor可以僅根據轉錄本的表達進行細胞分割,同時也支持與scRNA-seq獲得的細胞類型特異性表達譜的先驗信息整合,以及從共染色圖像中分割以改進分割。值得注意的是,Baysor使用馬爾可夫隨機場(MRF)來限制空間近端分子之間的關系。每個細胞都以高斯分布建模,整個數據集可以看作是細胞特異性分布的混合物,可以通過貝葉斯混合模型(Bayesian mixture models, bmm)進行分離。類似地,Sparcle利用Dirichlet過程混合模型進行初始細胞類型識別,并通過借用相鄰像素的信息,迭代地將每個轉錄本分配給細胞。另一種方法ClusterMap也利用鄰域的表達來計算鄰域基因組成,然后將細胞分割作為一個點模式分析問題,通過密度峰值聚類(DPC)算法求解。
在細胞分割后,可以像scRNA-seq一樣進行細胞水平分析,如差異表達分析和細胞-細胞相互作用。更重要的是,進一步探索細胞內的亞細胞結構成為可能。例如,在細胞分割的基礎上,ClusterMap可以使用K-means聚類進一步將細胞分割成包括細胞核和細胞質在內的亞細胞結構。Bento是一個用于ST數據亞細胞分析的工具包,它可以進一步識別RNA轉錄物的5類亞細胞定位,包括核、細胞質、核邊緣、細胞邊緣,以及以上都不是。
以上討論的細胞分割方法便于對基于成像的ST數據進行單細胞分析。然而,挑戰來自技術噪聲,如不均勻的強度信號和生物變異,包括不同的細胞大小和形狀以及不同的細胞密度。這些因素可能在實現準確的細胞分割方面造成困難,可能導致下游分析的偏差。因此,已經開發了幾種無分割方法,以便在不執行顯式分割的情況下進行穩健分析。大多數方法的目的是將每個分子像素分配給特定的細胞類型,而不是單個細胞。為了實現像素的細胞類型分配,Baysor的作者還提供了一種無分割的方法。它是基于相鄰RNA分子可能來自同一細胞的假設,共同反映了相應細胞類型的轉錄組學特征。他們為每個轉錄本計算一個鄰域組合向量(NCV),通過利用鄰域信息有效地增強一個像素的信號。ncv隨后被視為“偽細胞”,用于下游聚類和注釋分析。SSAM提供了一個類似的解決方案,它通過借用其鄰域的信息來估計每個像素的mRNA信號。不同的是,它們應用高斯核的核密度估計(KDE),這與Baysor不同,后者為考慮的最近鄰域提供相同的權重。
3. 整合scRNAseq解讀細胞類型的空間分布
無論組織樣本是單細胞還是空間轉錄組學,細胞類型注釋對于破譯細胞組成都是非常必要的。為scRNA-seq設計的注釋策略,包括基于表達標記基因的無監督聚類和細胞類型推斷,似乎適用于ST數據的分析。不幸的是,由于當前ST技術的限制,這種嘗試通常不會奏效。首先,對于基于成像的靶向ST技術,限制性的基因選擇和讀出噪聲的存在會阻礙未知細胞類型的鑒定。其次,對于基于條形碼的低分辨率ST數據,每個點的多個細胞或細胞類型混合的測量可能是平均的,這可能會模糊細胞的異質性。最后,對于基于條形碼的高分辨率ST數據,低檢測效率也對明顯聚類和適當的細胞類型標注提出了挑戰。因此,在大多數情況下,整合ST數據與匹配的scRNA-seq對于了解細胞類型分布是必要的。通常,積分可以通過兩種方法完成:映射或反卷積。細胞定位包括兩個方面:將預定義的細胞類型映射到空間位置和將scRNA-seq數據中的細胞映射到組織中。前者將細胞類型標記從scRNA-seq轉移到空間轉錄組學,后者預測來自scRNA-seq的細胞的空間位置,在某些情況下也被視為scRNA-seq的空間重構。對于細胞類型定位,可以使用來自scRNA-seq的細胞類型特異性基因特征來計算富集分數。該方法已被證明在分析基于微陣列的胰腺導管腺癌的ST數據方面是有效的。對于基因有限的基于成像的ST方法,上述的細胞分割方法,如pciSeq 、JSTA 和Baysor 也可以在scRNA-seq可用的情況下進行細胞類型分配。另外,由于這些基于成像的ST技術可以在細胞分割后提供單細胞水平的表達,因此現有的單細胞數據整合方法可以直接應用于單細胞分辨率空間數據和scRNA-seq的整合。例如,Seurat通過典型相關分析(CCA)將細胞從ST和scRNA-seq投射到共享潛在空間。以互近鄰(MNN)識別的細胞對為錨點,scRNA-seq的細胞類型標記可以轉移到空間細胞中。LIGER和Harmony也可以實現類似的集成。這些單細胞整合方法利用共同的潛在空間和鄰域信息,還可以預測ST缺失基因的空間表達,增強ST譜基因原有的弱信號。scRNA-seq的空間重建是在ST技術繁榮之前提出的,該技術通過一些空間地標性基因的表達來預測細胞的空間位置。早期的方法,如Seurat (v1.0),將含有數十個基因的ISH參考數據建模為二值化表達圖,然后通過將scRNA-seq衍生的雙峰混合模型與二值化表達參考相關聯,概率推斷出單個細胞的位置。Achim和DistMap也使用二值化的ISH參考,但采用不同的方法來計算細胞位置對應關系。Achim設計了一個評分方案,根據給定細胞中的基因特異性比率來評估細胞與每個空間位置之間的對應關系。
DistMap利用二值化的單細胞基因表達和空間參考計算馬修相關系數(Matthew correlation coefficient,MCC)得分,然后將細胞軟分配到空間位置。Tangram是最近開發的一種方法,除了基于ish的數據外,它還能夠將scRNA-seq與各種技術測量的空間轉錄組學相匹配。通過最大化scRNA-seq和ST共享的基因表達的相關性,Tangram可以得到一個概率映射矩陣,該矩陣表示在每個空間位置找到每個單個細胞的概率。最近的方法不是對細胞位置對應進行評分,而是將scRNAseq的空間重構問題轉化為監督學習問題或優化問題。例如,DEEPsc通過訓練基于神經網絡的分類器,將空間參考視為scRNA-seq,將細胞映射到空間位置的問題制定為監督分類問題。經過充分訓練的DEEPsc網絡將來自細胞的特征向量作為輸入,并根據來自不同空間位置的似然度預測細胞的空間起源。另一種方法,glmSMA將單元映射框架為凸優化問題。首先,采用拉普拉斯矩陣表示位置到位置的物理距離和細胞到細胞的表達距離。通過最小化每個細胞和相應位置的表達差異,glmSMA最終可以找到從scRNA-seq中的細胞到st中的空間位置的映射。SpaOTsc將細胞映射定義為一個最優運輸問題,旨在將細胞到位置的運輸成本最小化。SpaOTsc中的運輸成本主要基于scRNA-seq和空間參考之間的基因表達差異來衡量,并結合兩個懲罰項來處理兩個數據集的不平衡樣本量,并分別保留每個數據集內的結構。同樣,novoSpaRc采用最優轉運框架,其核心假設是物理近端細胞具有相似的表達譜。novoSpaRc通過位置到位置的物理距離和細胞到細胞的表達距離的組合來測量運輸成本,兩者都計算為各自kNN圖中的最短路徑。通過最小化運輸成本,novoSpaRc最終得到了一種映射,通過這種映射,細胞被映射到盡可能保留原始細胞-細胞對應關系的位置,考慮到上述假設。值得注意的是,novoSpaRc還允許在沒有參考ST數據時從頭重建scRNA-seq。大多數重建方法都是基于物理接近性可以通過表達相似性來反映的假設。然而,該假設不能代表所有細胞的空間分布模式,這使得推斷的細胞位置值得懷疑。
細胞類型反褶積旨在估計每個空間位置(即點或頭)的確切細胞類型比例,通常用于scRNA-seq和低分辨率條形碼的ST數據(如10x Visium)的整合。對于基于高分辨率條形碼的ST技術,如Stereo-seq,原始像素級表達式以基于bin的方式聚合,然后將每個bin作為一個新的空間單元進行反卷積分析。目前的ST反卷積方法基本上可以分為四類:回歸、因式分解、概率建模和基于圖的方法。回歸是一種最流行的方法開發的大量rna序列反褶積。由于每個點覆蓋的細胞數量有限,在ST數據上直接應用大量RNA-seq反卷積方法會導致來自不相關細胞類型的噪聲。為了克服這一問題,基于阻尼加權最小二乘(DWLS)回歸的ST反卷積方法spatialDWLS采用了兩種措施。首先,在精確估計細胞類型比例之前進行細胞類型富集分析,以確定每個點可能的細胞類型。其次,在對富集的細胞類型進行第一輪反褶積后,去除預測比例較低的細胞類型,進行另一輪反褶積。基于回歸的方法高度依賴于每種細胞類型的標記基因的選擇。與對細胞類型特異性表達譜進行回歸不同,一些方法提出對潛在主題譜進行回歸,該主題譜可以通過矩陣分解從單細胞表達數據中分解出來。例如,NMFreg最初是為Slide-seq的細胞類型注釋而開發的,它結合了非負矩陣分解(NMF)和非負最小二乘(NNLS)。它使用NMF從預標記的scRNA-seq中獲得一個基本的基因因子譜,然后使用NNLS回歸計算每個頭的因子負荷。由于每個因子都與細胞類型相關聯,因此因子負載充當細胞類型比例。SPOTlight采用了類似的策略,但使用了種子NMF,其中使用了細胞類型特異性標記基因和高可變基因(HVG)的組合,并通過scRNA-seq衍生的細胞-細胞類型歸屬初始化因子-細胞圖譜。反褶積也可以通過分解來實現。例如,STRIDE采用主題建模方法LDA,從scRNA-seq中獲得細胞類型相關的主題概況。然后,使用預訓練的主題模型可以推斷每個點的細胞類型組成。Stdeconvolve也基于LDA,但提供了一種無參考的解決方案。CARD建立在NMF的基礎上,但通過條件自回歸(CAR)模型考慮了點之間的空間相關性,這使得CARD成為一種“空間”反卷積方法。
除了直觀的回歸或基于因子分解的方法外,概率建模方法已經開發出來,假設細胞或點中的基因表達遵循特定的概率模型。例如,RCTD通過泊松分布對每個位置的基因表達進行建模,并將每個點擬合為單個細胞類型的線性組合。值得注意的是,RCTD還考慮了特定于平臺的效果。Cell2location遵循類似的概念,但使用NB分布來模擬基因表達。同樣,Stereoscope使用NB模型,但它適用于完整的基因集,而不是一組選定的標記基因。DestVI還使用NB分布來模擬每個基因在細胞或點中的表達,并使用神經網絡編碼和解碼參數。最重要的是,DestVI不僅可以估計細胞類型比例,還可以恢復每個點的細胞類型特異性表達,從而捕獲同一類型細胞內的連續表達變化。
除了DestVI之外,還有其他幾種基于神經網絡的方法。DSTG首先通過隨機混合scRNA-seq的細胞生成偽st數據,然后從偽st和實st構建跨點的鏈接圖。利用捕獲點之間內在拓撲相似性的鏈接圖,利用半監督圖卷積網絡(GCN)估計real-ST中每個點內的細胞類型比例。CellDART也生成偽st數據——一種虛擬的細胞混合物——但采用對抗性域適應的思想。CellDART集成了兩個基于神經網絡的分類器,其中訓練源分類器來預測細胞類型組成,訓練域分類器來區分真實斑點和偽斑點。通過在訓練過程中迭代更新兩個分類器,訓練良好的CellDART模型可以從真實ST數據中準確估計每個點的細胞類型比例。另一種基于神經網絡的方法GraphST采用了不同的策略。GraphST利用一個圖對比自監督框架,通過整合空間位置信息和本地上下文來重建ST數據的基因表達。使用自編碼器,GraphST可以單獨學習scRNA-seq的潛在表示。基于學習到的特征,通過對比學習機制訓練出細胞到點的映射概率矩陣,并結合scRNA-seq的細胞類型注釋提供對點的細胞類型組成。
4. 空間域識別
除了離散分布,我們還對細胞類型如何在空間上組織形成組織結構和執行功能感興趣。直觀地說,物理上近端的細胞,無論來自相同或不同的細胞類型,都可以構成一個空間結構,通常稱為空間域。空間域的識別將有助于我們理解區域內細胞之間的交流及其生物功能。從某種意義上說,空間域可以看作是具有特定空間模式的細胞群。scRNA-seq的標準Louvain聚類方法是基于基因表達相似性構建的圖,沒有考慮空間信息,在這里不直接適用。一些空間聚類方法改進了基于圖的聚類算法,以納入空間信息。例如,stLearn利用Louvain或K-means進行全局聚類,并通過考慮物理距離執行局部聚類來尋找空間分離的子聚類或合并空間近端單點。另一種方法是MULTILAYER,它在基因模式共表達圖上應用Louvain聚類。首先,MULTILAYER通過迭代凝聚策略檢測過表達基因的表達模式。基因表達模式在這里被定義為基因在多個連續位置過表達的區域。然后,MULTILAYER構建一個圖,其中節點表示先前檢測到的基因模式,邊緣表示基因模式之間的相似性(即基因共表達程度)。最后,利用Louvain算法將基因共表達模式劃分為多個組織群落。
許多空間聚類方法不是以間接方式合并空間信息,而是將空間接近信息編碼在MRF中,其中空間依賴關系由Potts模型表示。Zhu等人開發了smfishHmrf,該算法將隱馬爾可夫隨機場(HMRF)應用于seqFISH數據的空間域識別。他們首先構建一個鄰域圖來表示細胞之間的空間關系,其中馬爾可夫屬性只保留直接相鄰節點之間的關系。然后,他們通過聯合概率分布來建模每個細胞的區域狀態,該分布考慮了細胞的基因表達和鄰近細胞的區域狀態。通過使用期望最大化(EM)求解場平衡參數,smfishHmrf能夠檢測具有空間相干基因表達的空間域。BayesSpace采用帶MRF的全貝葉斯統計模型,以確保同一簇中的點在物理上更接近。BayesSpace通過使用Markov chain Monte Carlo (MCMC)和跨不同簇的固定精度矩陣,能夠穩定地估計模型參數,識別空間簇,甚至提高空間轉錄組學的分辨率。考慮到MCMC是計算密集型的,固定的平滑參數可能會限制不同ST數據集的性能,Yang等人提出了SC-MEB,以實現高效的計算和可調的平滑參數。特別地,他們采用了一種高效的基于迭代條件模型的期望最大化(ICM-EM)方案來估計參數,并通過改進的貝葉斯信息準則(MBIC)來選擇聚類數。上述基于磁共振成像的方法都假定隱藏的細胞狀態是離散的,這限制了我們對細胞間空間依賴性的理解。相比之下,SPICEMIX將NMF整合到HMRF中,其中觀察到的基因表達被建模為潛在因素的線性混合,潛在因素的混合權重被視為隱藏細胞狀態。SPICEMIX從另一個角度來理解,它提供了一種考慮空間信息的ST數據降維方法,可以作為下游聚類的基礎。基于推斷的細胞狀態,SPICEMIX進一步應用分層聚類來定義分類細胞類型。
在ST數據分析中,細胞類型聚類和空間域識別可以被視為兩個獨立的任務。我們上面討論的大多數方法都專注于識別空間域,除了SPICEMIX,其中空間聚類旨在推斷細胞類型,而不與scRNA-seq整合。另一種方法,FICT旨在通過空間聚類推斷基于fish的空間轉錄組學中的細胞類型。具體來說,FICT通過細胞類型特異性的高斯分布來模擬細胞的表達,并通過多項分布來模擬細胞與其相鄰細胞之間的關系。FICT能夠通過最大化聯合概率似然來分配單元簇。同樣,BASS也通過細胞類型特異性正態分布來模擬細胞中的基因表達,但同時,它通過特定域的分類分布來模擬細胞類型歸屬。有了這樣一個層次概率框架,BASS可以同時實現細胞類型聚類和空間域檢測。
空間轉錄組學可以看作是一個點圖,適合用于基于圖的神經網絡。許多基于gnn的方法已經被開發出來,通過整合基因表達和空間信息來學習空間轉錄組學的低維潛在表征,這可以促進下游分析,如空間域識別和空間變量基因的檢測。例如,SpaGCN應用GCN來整合多個信息源,包括基因表達、空間位置和組織學。首先,構建一個圖來表示點之間的關系,其中節點表示點,通過將組織學圖像特征轉換到第三個“z”坐標,并將其與點的原始空間坐標(x, y)結合,計算邊緣的距離。然后利用卷積層對圖中相鄰點的基因表達進行聚合。基于聚合的基因表達,實現無監督迭代聚類算法來識別聚類(即空間域)。
其他方法在基礎GCN中引入了額外的機制。正如我們在細胞型反卷積一節中討論的那樣,GraphST通過將基因表達與空間位置信息和本地上下文信息相結合,應用圖對比自監督框架來學習ST數據的空間潛在表示。另一種方法是SpaceFlow,它將深度圖信息集(DGI)框架集成到GCN編碼器中。除了基于空間轉錄組學構建的空間表達圖(SEG)外,SpaceFlow還通過隨機排列表達構建了表達排列圖(EPG)。這兩個圖都被送入圖卷積編碼器得到低維嵌入,DGI使編碼器通過判別器損失來區分SEG和EPG的嵌入。有些方法采用自編碼器進行空間嵌入。例如,SEDR使用深度自編碼器網絡來學習基因表達的低維潛在表示,然后使用變分圖自編碼器(VGAE)將其與空間信息集成。STAGATE為自編碼器引入了一種注意機制,使邊緣權重的自適應學習成為可能。例如,點相似度。stMVC構建了更全面的學習框架。具體來說,stMVC首先通過數據增強和對比學習,從組織學圖像中學習視覺特征。然后利用半監督圖注意自編碼器(SGATE)基于提取的視覺特征和空間基因表達獨立學習特定于視圖的表示,并通過注意機制整合兩個圖。stMVC提出的基于注意力的多視圖協同學習模型最終學習出一種更加魯棒的ST數據表示。由于ST數據的空間信號本質,一些方法將空間域識別問題轉化為經典的圖像分割問題。RESEPT使用GNN從點-點圖中學習三維嵌入,將基因表達作為節點的屬性,并通過邊緣連通性揭示物理鄰接性。將每個點的三維嵌入轉換為RGB尺度,使得之前為語義分割而設計的CNN可以直接應用于段空間域。另一種方法,Vesalius采用了類似的RGB嵌入策略,但通過UMAP而不是神經網絡進行降維。
5. 空間變異基因和基因表達模式的檢測
在scRNA-seq分析中,HVG在降維和隨后的細胞聚類中起著關鍵作用。在空間轉錄組學中,空間可變基因(SVG)的鑒定對于表征復雜組織中的功能組織也很重要。識別SVG就是尋找在空間上表現出很大變異的基因。scRNA-seq中的HVG檢測只考慮了高方差而忽略了空間信息,不能直接應用于SVG識別。已經提出了各種計算方法來從空間轉錄組學中檢測SVG。一些方法基于分割的空間域來識別SVG。例如,像我們上面討論的那樣,SpaGCN首先通過集成多個信息源來識別空間域,然后為每個識別的域定義相鄰域。空間可變基因是通過使用Wilcoxon秩和檢驗識別每個目標域和相應相鄰域之間的差異表達基因來確定的。大多數方法不依賴于空間域識別,而是直接將空間信息納入到模型中來研究基因表達的空間變異。根據核心模型,方法大致可分為三類:基于統計建模的方法、基于圖的方法和基于其他原理的方法。
1)基于統計建模
Trendsceek將空間表達式建模為標記點過程,其中空間位置被視為二維點過程,位置表達式被視為標記。對于給定的基因和指定的距離,對距離上的所有點對計算點的空間分布與其標記之間的依賴關系。依賴性評估可通過四項匯總統計來實現。Stoyan的標記-相關,均值-標記,方差-標記和標記-方差圖。當分數和分數的分布是獨立的時,匯總統計量將保持不變,但如果它們是相關的,則統計量將在不同的距離上變化。通過排列表達值來估計顯著性,不同距離間的p值最小即為該基因的顯著性。scGCO也利用標記點過程建模空間基因表達,但將HMRF集成到模型中。對于每個基因,scGCO通過圖切割算法對圖表示進行分割。在完全空間隨機框架下,這些片段可以作為候選區域來測試表達式對空間位置的依賴性,其中點在二維空間中的分布被建模為齊次泊松過程。
除標記點法外,還有許多方法采用高斯過程(GP)來模擬空間基因表達。GP是以時間或空間為索引的隨機變量集合,其中這些隨機變量的有限集合具有多元正態分布。GP在地質統計學中得到了廣泛的應用,并應用于空間轉錄組學建模。例如,SpatialDE基于高斯過程回歸,用空間和非空間方差項兩部分來模擬每個基因的可變性。可以通過計算這些項的比率來量化空間變異性(Svensson et al, 2018)。通過比較全模型與無空間協方差的零模型的似然,可以用對數似然檢驗估計統計顯著性。SpatialDE可以通過將擬合線性或周期(即余弦)協方差函數的完整模型與高斯核的模型進行比較,進一步識別具有不同類型空間變異的基因,包括線性或周期性模式。為了滿足高斯分布的假設,SpatialDE采用了兩步歸一化。具體來說,SpatialDE使用一種方差穩定變換方法,即Anscombe變換,對nb分布的原始計數進行變換,然后對對數總數進行回歸。Gpcounts也建立在高斯過程回歸的基礎上,但通過NB或零膨脹負二項(ZINB)分布而不是高斯分布來擬合空間計數。同樣,BOOST-GP通過ZINB分布來模擬基因讀取計數,但采用貝葉斯框架來推斷參數。另一種方法SPARK采用廣義線性空間模型(GLSM), GP建模空間位置之間的空間關系,泊松分布建模表達式計數數據。
此外,SPARK提供了一種更強大的統計方法來控制I類錯誤,它分別計算每個參數化核的p值,并將它們與Cauchy組合規則組合在一起。隨著ST技術的發展,需要對以往的方法進行改進,以適應高稀疏度的大規模空間轉錄組學數據。SpatialDE2基于SPARK,通過用omnibus test替代Cauchy combination,并引入Tensorflow的GPU加速來提高計算效率。為了降低計算復雜度和物理內存需求,SPARK的作者提出了一種可擴展的非參數測試方法SPARKX。具體來說,SPARK-X建立在一個非參數協方差測試框架之上,其中計算兩個協方差矩陣,分別測量表達相似性和空間接近性。然后將識別具有特定空間趨勢的基因轉化為檢測基因表達與空間位置的相關性。另一種方法是SOMDE,它將自組織映射(SOM)神經網絡整合到SpatialDE的高斯過程回歸框架中。SOMDE將原始空間位置濃縮為SOM節點,保留空間表達模式和拓撲結構。然后將原始空間表達聚合形成節點級基因元表達,顯著減小協方差矩陣的大小,從而提高計算效率。
2)基于圖表示
正如在空間域識別一節中所討論的,空間表達式可以用圖表示。一些基于圖形的方法已經被證明在SVG識別中是成功的。圖拉普拉斯分數通常用于基于圖的特征選擇,可用于從圖中識別空間變量基因。例如,GLISS首先建立一個相互最近鄰居圖,并計算每個基因的拉普拉斯分數,以測量其位置保持能力(即與局部結構的關聯)。在固定的圖中,低的拉普拉斯分數表明基因表達的相似性發生在較近的位置,而較大的差異發生在較遠的位置。每個基因的統計顯著性是通過排列表達和固定的圖來估計的。RayleighSelection提出了組合拉普拉斯分數,并將基于圖的表示擴展到空間表達數據的簡單復雜表示。除了圖中包含的頂點和邊,簡單復合體還包含高維元素,如三角形和四面體,可以捕獲更復雜的數據關系。因此,組合拉普拉斯分數有助于識別具有更復雜空間結構的基因。
有些方法在普通的圖表示中引入空間網格來簡化或優化空間結構。singleCellHaystack是一種基于空間網格的方法,最初用于預測從scRNA-seq學習的低維空間中差異表達的基因,獨立于細胞聚類。它還可以應用于使用自然二維或三維空間的空間轉錄組學數據的SVG識別。singleCellHaystack首先將多維空間劃分為網格,并定義網格點,用于估計空間中單元格的參考分布。然后,對于每個基因,singleCellHaystack根據二值化表達將所有細胞分為檢測組和未檢測組,并分別估計細胞分布。隨后計算Kullback-Leibler散度,通過與參考細胞分布的比較來衡量基因的散度,并通過排列檢驗來評估其顯著性。MERINGUE是另一種基于空間網格的方法。它首先使用Voronoi鑲嵌構造鄰域鄰接關系,Voronoi鑲嵌也用于構建scGCO中的圖表示。與k近鄰或k互近鄰相比,Voronoi鑲嵌可以適應不同的鄰域大小和距離,在不同細胞類型和非均勻密度的組織中具有更好的穩定性。然后,MERINGUE計算每個基因的Moran’s I來衡量空間自相關性,它表示空間相鄰位置之間的表達相關性。Giotto還提供了一種基于空間網格的方法BinSpect。類似地,BinSpect依靠Voronoi鑲嵌來確定鄰域關系。BinSpect采用統計富集分析,而不是Moran的I。對于每個基因,BinSpect使用k=2的k-means聚類或簡單的秩閾值對表達進行二值化。接下來,計算列聯表以反映相鄰位置之間的表達式依賴關系。然后采用Fisher精確檢驗來獲得優勢比和相應的p值。如果一個基因被發現是重要的,它往往在鄰近的位置高度表達。
3)基于其他原則
除了基于統計模型或圖形表示的方法之外,還有使用完全不同原理的方法。Sepal提出了一個獨特的策略擴散理論,將觀察到的基因表達譜視為轉錄物擴散的結果。在模擬的框架內,sepal假設轉錄本形成結構化模式比達到均勻隨機狀態需要更多的時間。因此,推斷基因表達模式的結構化程度轉化為測量模擬系統中的擴散時間。另一種方法,SPADE側重于識別與形態特征相關的重要基因。SPADE利用CNN從組織學圖像中提取潛在圖像特征。然后對高維特征進行主成分分析,總結圖像特征的空間分布規律。SPADE使用線性模型來發現與圖像模式(即pc)相關的基因,這些基因已被證明具有特定的空間趨勢。為了模擬基因表達的空間變異,上述方法只考慮位置之間的相對距離,而忽略了特定方向上的變異。SPATA為用戶提供了根據先驗知識手動定義軌跡軸的選項。對于每個基因,沿著預定義的空間軸擬合多個函數來模擬空間變化模式,包括線性,對數或梯度上升/下降,單峰或多峰函數。在所有函數中,通過對殘差求和的比較,選擇最適合的函數來表示基因的動態。在空間可變基因被識別后,一些方法通過聚類進一步確定原型基因模式。SpatialDE通過對聚類質心具有空間先驗的擴展高斯混合模型,對具有相似空間表達模式的svg進行聚類。
同樣,SPARK實現了一種分層聚類算法,將檢測到的變量基因分為不同的類別。MERINGUE不是基于表達構建相似性矩陣,而是通過計算空間相互關聯指數來推導相互關聯矩陣,這是對每對基因的Moran 's I自相關的改進。這構成了分層聚類的基礎。GLISS在潛在結構上擬合一個樣條模型,其中每個基因可以用擬合的樣條系數表示,具有相似基因模式的基因將共享相似的系數。與基于表達的相似度相比,基于樣條系數計算基因-基因相似度可以降低與空間變異無關的相關性。然后,GLISS對系數進行譜聚類,將基因聚類成組。
6. 偽時間軌跡分析
從scRNA-seq或ST數據中,我們只捕獲細胞基因表達的快照。通過以上的空間域檢測或SVG識別,我們可以分別以離散或連續的方式研究空間上的轉錄動態。之前在scRNA-seq偽時間分析方面的努力為我們提供了僅從表達數據重建細胞狀態軌跡的機會。ST帶來的附加空間信息通過引入空間維度擴展了原有的偽時間分析。在ST數據上直接應用單細胞偽時間方法可能導致細胞軌跡隨時間連續而在空間上不連續。為了解決這個問題,stLearn通過加入空間信息來調整原始的偽時間算法。stLearn首先利用擴散偽時間(DPT)算法從基因表達中預測偽時間。然后結合基于表達式的偽時間和空間距離的差異計算偽時空距離(PSTD)矩陣,并用一個權值來平衡它們。基于PSTD矩陣,stLearn構建了一個有向圖,并應用最小生成樹算法來確定分支(即推斷細胞軌跡)。與其依賴于僅從基因表達推斷的初始偽時間軌跡,還出現了幾種從組合表達和空間信息預測細胞軌跡的方法。在空間域識別一節中討論了SpaceFlow,它提供了一個深度學習框架,可以從ST數據中學習低維嵌入。SpaceFlow生成的嵌入可用于利用DPT算法計算偽時空圖(pseudo-Spatiotemporal Map, pSM),便于從ST數據中綜合重建時空軌跡。因此,SpaceFlow生成的時空順序在空間和偽時間上都保持一致性。
7. 細胞-細胞通訊和基因-基因相互作用
通過上述分析,我們可以對細胞類型的空間分布和表達的空間變化有一個基本的了解。然而,細胞或細胞類型的組織,以及產生這種空間模式的基因調控,仍然難以捉摸。許多研究報道,細胞行為可以由來自環境的細胞信號通路塑造。空間轉錄組學提供了一個獨特的機會來研究保存微環境中的細胞-細胞通訊。利用ST數據探索細胞間空間依賴性的方法有幾種,其中最直觀的方法是研究不同細胞類型的鄰近或共定位。例如,Giotto采用隨機排列策略來識別富集的細胞型對。在鄰域網絡結構固定的情況下,對節點間的細胞類型標簽進行洗牌,形成隨機的鄰域關系。通過這種方式,可以確定兩種細胞類型之間觀察到的超期望頻率的比率,并可以估計相應的富集顯著性。spicyR最初是為原位細胞術的空間分析而設計的,它定義了一個分數來衡量細胞類型共定位的程度。spicyR通過標記點過程模擬細胞的空間分布,應用k函數或方差穩定的k函數(即l函數)來量化特定距離內兩種細胞類型之間的共定位。最近,ang等人基于集體最優運輸方法開發了COMMOT,用于處理復雜的分子相互作用和空間約束,以推斷空間分辨轉錄組學中旁分泌依賴的細胞-細胞通信。
除了觀察到的細胞類型的共定位外,細胞之間的空間依賴關系可能更為復雜,需要通過更復雜的方法來建模。NCEM在以節點為中心的表達模型中協調方差歸因和細胞-細胞通信。NCEM首先使用圖結構對單元通信實施鄰域約束。通過提供的細胞類型標簽,NCEM根據細胞類型和空間背景應用一個功能來擬合細胞所觀察到的基因表達。為了適應不同場景中空間依賴關系的復雜性,NCEM提供了三個模型,包括線性、非線性和生成潛變量模型,分別由線性回歸、非線性編碼器-解碼器GNN和條件變分自編碼器實現。通過對目標細胞(即接收器)的分子狀態對鄰域(即發送方)的依賴性進行建模,NCEM還可以確定發送方-接收器信號的方向性。不同于對整個表達譜依賴于細胞間通訊的建模,有幾種方法量化了細胞間相互作用對每個基因表達的影響。例如,SVCA使用高斯過程模型對靶基因在細胞間的表達進行建模,并將基因的變異性分解為三個組成部分,包括內在效應、來自未測量空間變量的環境效應以及來自鄰近細胞的細胞間相互作用效應。通過這種方式,可以估計每個基因的每個術語解釋的方差的比例,并且可以識別參與細胞-細胞相互作用的生物學相關基因。MISTy設計了一個多視圖框架來解釋單個基因的表達,其中來自不同空間背景的細胞-細胞相互作用在不同的視圖中建模。與SVCA相似,MISTy包括內觀、近觀和旁觀,分別對應于同一位置其他基因表達的內在影響、近鄰的影響和組織結構(即指定細胞半徑內的細胞)的影響。通過分析每個預測基因在每個視圖中對目標基因的重要性,不同空間背景的影響可以解釋感興趣的基因對。SVCA和MISTy可以模擬基因-基因關系,發現與細胞-細胞相互作用相關的基因,但它們都不能識別顯性基因-基因相互作用對。Yuan和Bar-Joseph開發了GCNG,一種基于gcns的監督計算框架,用于預測基因相互作用。GCNG以空間鄰域的圖表示作為輸入,以及候選基因對的歸一化表達。輸出將是相互作用或非相互作用基因對的分類。
為了實現監督學習,已知的配體-受體相互作用被標記為正對,隨機選擇的配體-受體對被標記為負數據。GCNG具有五層GCN結構,可以預測所研究的ST數據集中新的基因-基因相互作用。然而,GCNG不能告知相互作用發生的細胞類型,也不能關注特定局部區域內的相互作用推斷。為了解決這些局限性,一些方法通過考慮細胞類型位置依賴于配體和受體的共表達。例如,MERINGUE進一步將基因對之間的空間互相關計算限制為篩選的配體-受體對和兩種感興趣的細胞類型。Garcia-Alonso等人將他們的CellphoneDB升級到v3.0,可以在特定的微環境中識別感興趣的細胞類型的配體-受體對。同樣,在上一步細胞類型接近分析的基礎上,Giotto通過計算相互作用細胞類型的細胞亞群中配體和受體的加權平均表達來定義配體-受體相互作用評分。
8. 空間數據綜合分析
隨著通量的增加和成本的降低,一些研究從多個個體中生成ST幻燈片以進行大規模分析。其他一些研究從組織的多個相鄰層產生一系列ST玻片,從而實現整個組織的全局視圖。對單個ST玻片進行單獨分析可能會降低多個樣品的功效。因此,需要采用積分方法對多個樣本進行聯合分析。此外,隨著形態學等附加信息的提供,空間轉錄組學應該與其他模式相結合,以全面表征組織。在本節中,我們將回顧多樣本集成和不同模態空間數據集成的計算方法。
1)多試樣集成
多樣本積分的核心是將多個樣本放置在同一空間,稱為共同坐標框架(common coordinate framework, CCF)。坐標系包含兩個方面。一方面,CCF可以代表自然的三維空間,其中多個平面幻燈片排列堆疊,提供組織的立體視圖。另一方面,來自多個樣本的高維空間位置測量可以投影到共享的低維空間中,用于聯合空間域識別等綜合分析。
已經開發了一些方法來排列來自同一組織的多個連續載玻片。PASTE將多片排列表述為一個最優運輸問題,該問題基于基因表達和空間信息計算概率排列。通過最小化運輸成本,PASTE可以實現最大限度地提高幻燈片上對齊位置之間的基因表達相似性,同時保留幻燈片內的空間結構。PASTE可以對齊來自同一組織的多個連續幻燈片,但不能應用于來自不同時間點的幻燈片的集成。Andersson等人提出了一種方法eggplant,這是一種基于地標的方法,將多個幻燈片投影到共同參考文獻中。首先,eggplant將測量到的空間位置投影到參考點上,使變換前后地標之間的距離保持不變。接下來,eggplant應用高斯過程回歸來學習所有排除地標的位置的基因表達與到地標的距離之間的關系,從而可以預測參考中每個位置的基因表達。采用位置轉換與表達預測相結合的策略,可以將不同時間點或不同個體的多張幻燈片轉移到同一參考文獻中進行綜合分析。然而,eggplant不僅需要選擇地標位置,還需要定義參考,參考通常是代表組織域的規范結構。這兩個要求限制了eggplant在腫瘤等更復雜組織上的應用。為了解決這個問題,Jones等人開發了GPSA,它也是基于高斯過程模型。GPSA構建了一個雙層高斯過程框架,其中第一層將測量的空間位置映射到一個公共坐標系,第二層描述該系統內的空間基因表達。與eggplant相比,GPSA可以從頭迭代估計公共坐標系統,但它也提供了基于模板的與預定義的公共坐標對齊的選項系統通過固定一個幻燈片。
不同于將空間位置從多張幻燈片映射到自然3D空間中的CCF,有幾種方法側重于將多個樣本投影到共享的低維空間。在這種情況下,整合方法應該能夠從不同批次中去除不需要的變異,并保留有意義的生物變異,如scRNA-seq。但不同于單細胞積分法,ST積分法需要考慮空間信息。Liu等人提出了PRECAST,這是一種統一的原則性概率模型,用于聯合估計低維嵌入并在多個組織載片上執行空間聚類。PRECAST采用內稟條件自回歸(CAR)模型對歸一化的基因表達進行降維,在低維空間中保持了鄰居之間原有的空間依賴性。由此產生的潛在低維嵌入可以進一步利用HMRF模型進行空間聚類。正如我們上面提到的,BASS支持多尺度分析,同時進行細胞類型聚類和空間域檢測。它還允許多樣本整合分析,通過聯合建模和諧校正的空間轉錄組與層次貝葉斯框架。另一種方法,MAPLE提出了一個混合框架,用于多個部分的聯合空間聚類,通過基于gcn的模型進行空間感知的低維嵌入學習。
2)多模態集成
如上所述,單細胞和空間轉錄組學通常結合起來,通過細胞作圖或細胞類型反褶積來破譯細胞類型的空間分布。在我們回顧的整合方法中,Tangram脫穎而出,通過與多模態單細胞數據集成,將其他模式的數據映射到空間轉錄組學。例如,一旦SHAREseq的單細胞通過基因表達相似性定位到空間位置,染色質可及性的空間模式就可以被揭示。考慮到許多ST技術提供相應的組織學圖像,許多計算方法利用額外的圖像信息來提高每一步的分析性能。例如,stLearn利用形態相似性對表達數據進行規范化,從而減少技術噪聲的影響。在計算點-點距離構建空間轉錄組學圖時,spaGCN考慮了組織學圖像特征。stMVC采用帶有注意機制的圖網絡來整合包括組織學特征在內的多源信息,最終學習ST數據的低維嵌入。同樣,conST和MUSE等方法也使用深度學習架構來整合細胞形態和轉錄狀態以進行聯合表示。SPADE沒有采用復雜的基于深度學習的機制,而是使用線性回歸模型直接將基因表達的空間方差與圖像特征的空間分布模式聯系起來。
除了便于空間轉錄組學分析外,組織學圖像還可用于預測空間基因表達。已經開發了許多方法來解決這個問題。為了克服一些基于條形碼的ST技術的低分辨率限制,bergenstratuhle等人提出了一種深度生成模型,從高分辨率組織學圖像中推斷超分辨率表達圖,包括原始測量位置內部和之間。一些方法不是專注于提高空間基因表達的分辨率,而是將空間轉錄組預測推廣到沒有匹配表達數據的組織病理圖像。例如,He等人引入了一種深度學習算法ST-Net,通過結合空間轉錄組學和組織學圖像來捕獲基因表達異質性。該模型使用包含68個乳腺組織切片的ST切片的BRCA空間轉錄組數據集進行訓練,可以直接從組織學圖像中預測其他乳腺癌數據集的空間分辨率轉錄組。然而,ST-Net并沒有考慮到點之間的空間依賴性。HisToGene采用了一種改進的Vision Transformer模型來預測空間基因表達,并考慮了位點依賴性。在HisToGene的基礎上,Hist2ST還包括一個Convmixer模塊,用于捕獲圖像斑塊內2D視覺特征的內部關系。
應用程序
近年來空間轉錄組學的快速發展促進了其在各種生物系統中的廣泛應用。ST技術在空間表征健康組織的細胞狀態方面發揮了重要作用,其中一些技術旨在破譯特定發育階段組織的空間結構。值得注意的是,在這些組織中,神經系統一直是研究的焦點。許多研究為構建詳細的大腦空間圖譜做出了重大貢獻。此外,ST技術在探索損傷或病變組織的微環境方面已被證明是無價的,包括感染病毒的小鼠肺部,心肌梗死的人類心臟以及一系列不同的腫瘤類型。本文綜述了ST在三個主要領域的應用,包括健康組織的發育和動態平衡、神經科學和腫瘤微環境。
1)健康組織的發育和體內平衡
大多數研究利用小鼠模型來研究早期哺乳動物胚胎的發育。已經建立了小鼠胚胎發育的幾個階段的空間圖譜。Peng等人關注著床后階段的譜系分化和形態發生。Geo-seq應用于從原腸形成前(胚胎期第I5.5天)到原腸形成后期(胚胎期第7.5天)所有胚層中預先選擇位置的細胞群。該研究揭示了譜系規范和組織模式在時間和空間上的動態分子調控。此外,他們還發現了Hippo/Yap信號在細菌層發育過程中的關鍵作用。為了進一步探索原腸胚形成結束時早期器官發生中細胞命運的決定,Lohoff等對E8.5 - E8.75時收集的小鼠胚胎的多個矢狀切片進行了seqFISH。由于目標基因數量有限,他們將seqFISH與現有的單細胞轉錄組圖譜整合,以實現全基因組的植入。利用生成的單細胞空間圖譜,揭示了早期發現的中腦和后腦區域背-腹軸和喙-尾軸對應的基因表達空間模式腸管背腹分離。最近,Chen等人將高分辨率Stereo-seq技術應用于E9.5 ~ E16.5妊娠中后期的全鼠胚胎,最終構建了小鼠器官發生時空轉錄組圖譜(MOSTA)。除了小鼠的早期胚胎發育,許多研究人員已經利用空間轉錄組學來探索驅動人類器官或組織發育的空間依賴機制。例如,Crosse等人利用基于lcm的RNA測序技術,對卡內基階段(CS)16 - CS17(即受孕后39-41天)的人類胚胎中正在發育的造血干細胞(HSC)生態位進行了空間分辨分析。他們分析了主動脈的背腹側極化信號,并確定腹側分泌的內皮素是早期人類HSC發育的重要分泌調節因子。在對人類心臟發育的研究中,Asp等人使用ST技術表征了人類心臟在三個發育階段(受孕后4.5 - 5周、6.5周和9周)的不同解剖區域。通過scRNA-seq和ISS的整合,建立了一個全面的空間圖譜,提供了人類心臟發生過程中細胞亞型定位的詳細信息。類似的策略也應用于8至22 PCW的人類腸道發育研究。除了生成人類腸道發育的時空圖譜外,他們還揭示了形態梯度如何指導細胞分化。空間轉錄組學也被應用于成人健康組織的細胞型圖譜和穩態維持的研究,可以作為與病變組織比較的參考。Shen等利用Stereo-seq技術繪制了人類牙齦的ST圖譜。通過鑒定牙周炎相關效應細胞、基因和途徑,ST結果可能有助于開發新的牙周炎治療策略。Madissoon等人通過結合scRNA-seq、snRNA-seq和10x Visium ST,創建了人類肺和氣道的空間多組學圖譜,其中包括各種新的和已知的細胞類型。空間肺圖譜還揭示了特定的組織微環境,如腺體相關淋巴細胞生態位(GALN),可能在預防呼吸道感染中發揮作用。在另一項關于人類子宮的研究中,Garcia-Alonso等也應用多組學技術構建了人類子宮內膜的綜合細胞圖譜,表征了整個月經周期的時空動態。特別是,進一步的空間相互作用分析揭示了NOTCH和WNT信號通路在塑造纖毛和分泌細胞系分化中的作用。隨著ST數據的積累,可以預見,在不久的將來,多源組織圖的整合將導致整個人體綜合參考空間圖譜的建立。
2)神經科學
明確的分層結構和獨特的解剖區域使大腦成為驗證新開發的空間轉錄組學技術的合適材料。反過來,這些ST技術顯著增強了我們對大腦空間結構的理解。許多研究都致力于構建大腦的參考圖。由于早期基于成像的ST技術的視野大小有限和密集勞動性質,大多數研究集中在小鼠大腦的特定亞區域。例如,Codeluppi等人開發了osmFISH,并使用該方法定義了體感覺皮層的空間細胞組織,僅涵蓋33個靶向標記基因和約5000個細胞。與此同時,Moffitt等人通過將MERFISH與scRNA-seq相結合,生成了下丘腦視前區神經元的空間分子圖譜。同樣,大腦的其他亞區,如視覺皮層、初級運動皮層、海馬和小腦,已經通過不同的基于成像的ST技術來建立詳細的空間細胞組織圖。由于基于高通量條形碼的ST技術的發展,Ortiz等人建立了整個成年小鼠大腦的分子圖譜。他們利用ST技術分析了從一個大腦半球沿前后軸收集的75個鄰近冠狀面切片的空間基因表達。通過與Allen小鼠腦圖譜(ABA)的比對,他們構建了一個完整的腦圖譜,提供了三維組織坐標和詳細的ABA神經解剖學定義。更重要的是,他們還通過無監督分類在分子圖譜中定義了新的區域和層特異性亞區。無論是整個大腦還是特定的子區域,這些圖譜對實驗神經科學都有很大的價值,最終擴展了我們對大腦結構-功能關系的認識。
除了揭示正常大腦中細胞類型的空間組織外,空間轉錄組學還可以擴展到神經退行性疾病或精神疾病的研究,揭示神經系統功能障礙或失調的空間相關機制。例如,Chen等將ST技術與ISS相結合,捕捉了阿爾茨海默病(AD)淀粉樣斑塊附近的轉錄變化。特別是,他們確定了兩個基因共表達網絡,可能對阿爾茨海默病中的淀粉樣斑塊沉積有反應。Maniatis等在肌萎縮性側索硬化癥(ALS)的研究中,利用不同階段的ALS小鼠模型,采用ST技術表征疾病進展的時空動態。結合ALS患者的死后組織,他們在與ALS病理相關的轉錄途徑中發現了共同的空間擾動模式。隨著ST技術在分辨率和檢測效率方面的不斷提高,我們期望建立更詳細和全面的神經系統地圖集。這些資源對于探索電路和行為的結構-功能關系無疑是無價的。
3)腫瘤微環境
盡管單細胞轉錄組學已經揭示了復雜的TME中的細胞類型組成及其功能,但仍未探索這些細胞如何在空間上組織以控制或促進腫瘤進展。空間轉錄組學使研究不同的細胞群體和信號通路與空間背景保存成為可能。腫瘤微環境一般包括腫瘤細胞、基質細胞和免疫細胞。最初的研究工作往往集中在腫瘤區域的內部異質性。在人類乳腺癌(BRCA)的單細胞和空間圖譜研究中,Wu等人從scRNA-seq中衍生出7個基因模塊來描述腫瘤內的轉錄異質性。分析發現兩個在腫瘤區域相互排斥的基因模塊,它們分別與EMT和增殖狀態有關。在另一項原發性肝癌的研究中,定義了5種癌癥干細胞(cancer stem cell,CSC)群體,它們在不同的區域表現出不同的分布模式,包括前沿、腫瘤和高級別門靜脈腫瘤血栓形成。值得注意的是,PROM1+ CSCs在門靜脈腫瘤血栓中的比例高于腫瘤區域,可能在腫瘤進展中發揮關鍵作用。
以腫瘤區域為中心,利用空間轉錄組學可以揭示免疫或基質細胞類型的相對空間分布。Ji等在人鱗狀細胞癌(SCC)的研究中發現,B細胞在腫瘤中浸潤,而在腫瘤-間質邊界大量存在調節性T細胞、巨噬細胞和成纖維細胞。相反,CD8 T細胞明顯被排除在腫瘤之外。同樣,不同的細胞亞型或狀態也顯示出不同的空間模式。Wu等在乳腺癌的TME中發現了炎癥樣癌癥相關成纖維細胞(iCAFs)和肌成纖維細胞樣CAFs (mCAFs),但這兩種亞型表現出明顯不同的空間分布。mCAFs富集于浸潤性癌區,而iCAFs分散分布于浸潤性癌區、間質區和淋巴細胞聚集區。一些研究感興趣的是腫瘤-基質邊界的分子和細胞類型模式,即腫瘤的侵襲。Wu等人描述了侵襲中細胞類型豐度的動態變化,并在邊界附近區域發現了免疫抑制微環境。
空間分析還可以識別腫瘤微環境中的一些模式結構并對其進行表征。在上述肝癌研究中,對ST點進行無監督聚類,發現了一個以三級淋巴結構(TLS)相關基因高表達為特征的聚類,如CXCL13、CCL19、CCL21、LTF和LTB。病理檢查證實存在TLSs。Wu等定義了一種TLS-50特征來定位其他組織切片中的tls,也發現TCGA的HCC患者預后較好。同樣,Andersson等也在her2陽性乳腺癌中發現了TLSs。為了進一步研究TLS如何影響癌癥對免疫治療的反應,梅蘭等人使用空間轉錄組學研究了腎細胞癌(RCC)中TLS內B細胞反應的性質。他們發現TLSs可以產生和繁殖產生抗腫瘤抗體的漿細胞,這與免疫治療的反應有關。已知細胞通訊在腫瘤的免疫監視或逃逸以及腫瘤進展中發揮重要作用。通過細胞類型反褶積或細胞作圖分析揭示細胞類型的空間分布,還可以識別細胞類型接近或共定位模式。
Moncada等人通過將scrna -seq定義的細胞類型定位到胰腺導管腺癌的ST,確定了炎癥成纖維細胞和應激反應癌細胞的共定位。同樣,scRNA-seq和ST在SCC中的整合,在腫瘤特異性角化細胞群周圍發現了纖維血管生態位。進一步的相互作用分析表明,共定位可能由多個配體-受體對介導。在結直腸癌的另一項研究中,空間轉錄組學和免疫熒光染色顯示FAP+成纖維細胞和SPP1+巨噬細胞共存,這與患者生存率低有關。
隨著空間多組學技術的發展,細胞串擾和代謝狀態等其他方面的特征將被表征,以獲得對腫瘤微環境復雜性的更多見解。了解腫瘤微環境有助于確定治療靶點和設計抗腫瘤藥物。
總結
本章全面概述了當前空間轉錄組學的進展,包括技術創新、計算方法和各種應用。空間轉錄組學徹底改變了我們對組織組織和細胞異質性的理解,使完整組織內基因表達模式的高分辨率可視化成為可能。計算方法的發展促進了空間轉錄組學數據的整合和解釋,揭示了空間調控機制和新的分子相互作用。空間轉錄組學已經成功地應用于各個領域,包括發育生物學、神經科學、癌癥研究和免疫學,具有加速生物標志物發現和個性化醫學方法的潛力。空間轉錄組學代表了一種變革性的方法,并將繼續完善以重塑我們對復雜生物系統的理解。我們預計它將為組織穩態和疾病機制提供深刻的見解。
參考文獻:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0