單細胞轉(zhuǎn)錄組:聚類分析中的機器學(xué)習(xí)與統(tǒng)計方法綜述(三)
接上回,在單細胞轉(zhuǎn)錄組 | 聚類分析中的機器學(xué)習(xí)與統(tǒng)計方法綜述(一)和單細胞轉(zhuǎn)錄組 | 聚類分析中的機器學(xué)習(xí)與統(tǒng)計方法綜述(二)中,綜述了在過去幾年間發(fā)展起來的,用于單細胞轉(zhuǎn)錄組分析中聚類的機器學(xué)習(xí)和統(tǒng)計方法,重點介紹了如何將一些常見的聚類方法,如層次聚類、基于圖的聚類、混合模型、k-means、集成學(xué)習(xí)、神經(jīng)網(wǎng)絡(luò)和基于密度的聚類等加以調(diào)整及應(yīng)用,從而解決單細胞轉(zhuǎn)錄組數(shù)據(jù)分析中的獨特挑戰(zhàn),例如低表達基因的缺失,轉(zhuǎn)錄本的不均勻覆蓋,以及由技術(shù)偏差和不相關(guān)的混雜生物變異所帶來的細胞標(biāo)記的失真。我們評價了標(biāo)準化、dropouts推測以及降維等預(yù)處理步驟如何提高聚類效果。
本文將繼續(xù)介紹一些能夠?qū)r間序列樣本和多個細胞群進行聚類并且檢測罕見細胞類型的新方法。最后,對部分開發(fā)用于單細胞轉(zhuǎn)錄組聚類分析的軟件進行了實驗和比較,以評估其性能和效率,為未來的數(shù)據(jù)分析提供一定的指導(dǎo)和方向。
01 罕見細胞類型及單個細胞類群
在單細胞的聚類分析中,罕見細胞類型的檢測是一個重要問題,因為在發(fā)育或疾病進展中起重要作用的細胞類型往往豐度較低。由于罕見細胞類型的群體規(guī)模小,在標(biāo)準的聚類分析中往往難以檢測到。
RaceID是專門用于從單細胞轉(zhuǎn)錄組數(shù)據(jù)中識別稀有細胞類型的一種聚類工具。該工具首先計算細胞間Pearson相關(guān)性用于k-means聚類。在每個類群中,根據(jù)和背景噪聲模型相比的基因的變異型篩選離群細胞。最后,如果離群細胞的相關(guān)性超過原始聚類中細胞間相關(guān)性的閾值,則將離群細胞合并到離群簇中。
GiniClust是另一個聚焦于罕見細胞發(fā)現(xiàn)的聚類工具。在算法中使用基尼系數(shù)進行特征基因的選擇。與常用的Fano因子相比,這種方法對細胞總量占比較低的細胞群體更加敏感。最后,利用基尼系數(shù)篩選得到的基因作為特征進行DBSCAN密度聚類,檢測稀有細胞類型。
屬于稀有細胞類型的細胞也可以被視為聚類過程中產(chǎn)生的異常值。在大多數(shù)已公開的單細胞聚類算法中,都可以生成數(shù)量相對較小的簇,甚至該簇中只包含一個細胞。雖然這可能是由于聚類算法的初始化或收斂性差造成的,但它也可以被解釋為來自罕見細胞類型的異常細胞。一些算法或工具包含特定的技術(shù)和參數(shù),能夠進行罕見細胞類型的檢測。以使用層次聚類的SINCERA為例,它不要求用戶指定簇之間的最小距離,而是使用允許的最低細胞數(shù)量的閾值。
02 細胞Marker基因的檢測
聚類分析的另一個重要目的是發(fā)現(xiàn)新的Marker基因,以描述通過聚類發(fā)現(xiàn)的每種細胞類型的基因表達模式和功能,從而用于未來的生物學(xué)解釋和實驗驗證。大多數(shù)方法是在聚類后通過對不同類群之間的差異表達基因進行統(tǒng)計檢驗分析來識別Marker。例如,Seurat使用Wilcoxon秩和檢驗,這是一種基于排序表達值中秩次統(tǒng)計量的非參數(shù)檢驗方法。在SINCERA中,當(dāng)樣本容量較小,同樣使用秩和檢驗,當(dāng)樣本容量變大時則使用Welch’s t檢驗。
除了上述方法是將差異表達分析作為聚類的后處理步驟,還有一些則是在聚類的過程中同時進行Marker基因的檢測。BackSPIN計算每次分裂后每個簇中的平均基因表達量,并將每個基因分配到表達量最高的簇中。DendroSplit通過Welch’s t檢驗識別p值最顯著的Marker基因作為類群分離評分,以決定是否需要在層次聚類中進一步拆分分支。SAIC使用k-means對細胞進行聚類的同時,利用方差分析選擇Marker基因。
03 方法評估
在本節(jié)中,我們對單細胞轉(zhuǎn)錄組的聚類方法進行了兩次實驗評估。在第一個實驗中,我們使用人外周血單細胞數(shù)據(jù),比較了幾種廣泛使用的單細胞聚類工具或方法,以確定不同方法的優(yōu)勢和局限性。在第二個實驗中,我們對來自5個個體的212個乳腺癌細胞進行了聚類,以評估不同的工具在不同批次來源的多個細胞群中的聚類性能。
● 人外周血數(shù)據(jù)
我們從10x Genomics網(wǎng)站下載了PBMC數(shù)據(jù),在原始數(shù)據(jù)中總共包含了103887個細胞。除了使用整個數(shù)據(jù)集去進行方法的比較外,我們還對原始數(shù)據(jù)按照不同大小(100,1000,10000)進行向下采樣以評估其延展性。數(shù)據(jù)集最初包含32739個表達基因,我們從中選擇了至少在3個細胞中表達的19630個基因。(使用的計算機參數(shù):Intel Xeon E52687W v3 3.10GHz, 25 M Cache and 256 GB of RAM)。

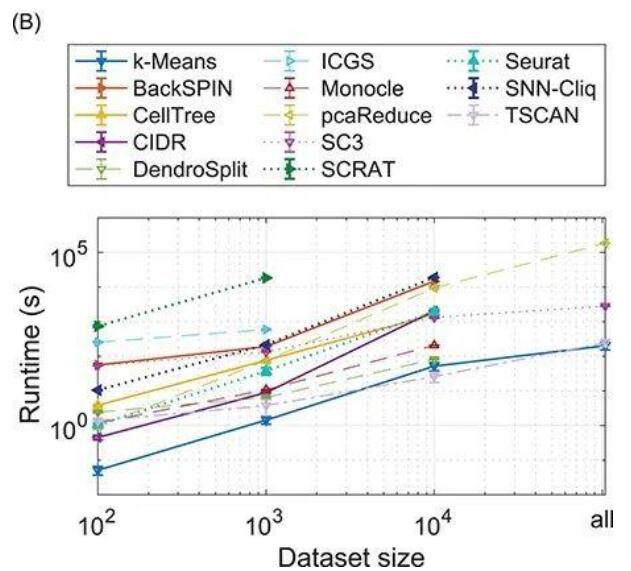
Figure 5. PBMC中聚類方法的比較
(A)Y軸表示ARI值,X軸表示不同的測試數(shù)據(jù)集。其中不同顏色代表了不同的工具或方法。(B)Y軸表示運行時間,X軸同圖A。曲線截斷表明該方法在相應(yīng)數(shù)據(jù)集下不再適用。
如圖5所示,通過對10次不同的運行結(jié)果取均值和標(biāo)準差,對ARI和運行時間進行了比較。結(jié)果表明,在這些方法中,Monocle、cellTree、Seurat和SC3的表現(xiàn)最好。但是,由于內(nèi)存問題,Monocle、cellTree和Seurat不能擴展到所有的測試數(shù)據(jù)集。SC3的算法中,最多對5000個細胞進行聚類,剩余的細胞則通過構(gòu)造一個支持向量機(SVM)完成。而除去這一監(jiān)督學(xué)習(xí)的步驟,SC3的表現(xiàn)和cellTree、Seurat相似。pcaReduce能夠應(yīng)用于所有的數(shù)據(jù)集,但運行時間超過2天(圖5B),同時聚類結(jié)果并沒有因為數(shù)據(jù)集包含細胞數(shù)的增多而得到改善(圖5A)。SCRAT包在對100個細胞時進行聚類時表現(xiàn)良好,但當(dāng)使用40個單元(此處單元表示具有相關(guān)基因表達的細胞)聚類1000個細胞時變得不穩(wěn)定。此外,該工具至少需要3天時間來處理5000個細胞的數(shù)據(jù)集,因此不能擴展到更大的數(shù)據(jù)集。
圖5A還顯示,SC3和pcaReduce等使用k-means作為聚類步驟之一的工具在多次運行中的方差最大,而使用層次聚類的工具cellTree、CIDR和DendroSplit,使用基于圖聚類方法的工具SNN-Cliq和基于密度的聚類工具Monocle在多次運行中總是保持相同的聚類結(jié)果。混合模型TSCAN和Seurat以及神經(jīng)網(wǎng)絡(luò)方法SCRAT也返回相同的聚類結(jié)果,這表明在聚類實現(xiàn)的過程中使用了一些固定的初始化策略。
進一步分析發(fā)現(xiàn),基于層次聚類的方法顯示出非常接近的平均ARI結(jié)果。當(dāng)聚類1000個細胞時,我們可以看到BackSPIN、CIDR、DendroSplit和ICGS的ARI值大約在0.25到0.3之間。cellTree雖然也是基于層次聚類,但應(yīng)用了LDA對數(shù)據(jù)進行降維,這似乎更適用于原始計數(shù)數(shù)據(jù)。在基于劃分的聚類方法上,我們可以看到,盡管pcaReduce使用k-means作為其框架的一部分,但通過正確使用PCA和聚類的合并策略,能夠顯著改善聚類的結(jié)果。SC3看起來是一種前景不錯的方法,它結(jié)合了幾種不同的距離測量和映射方法的優(yōu)點,然而,當(dāng)數(shù)據(jù)集增大,即SC3開始依賴SVM對更多的細胞進行分類時,結(jié)果似乎是不穩(wěn)定的,例如聚類10000個細胞的結(jié)果要差于聚類1000個細胞的結(jié)果。使用GMM的TSCAN在大數(shù)據(jù)集中表現(xiàn)出比k-means更好的結(jié)果,這表明高斯混合模型可能在聚類中發(fā)揮更好地積極推動作用。對于基于密度的聚類,Monocle在聚類10000個細胞時的性能優(yōu)于其他方法。最后,盡管Seurat和SNN-Cliq都建立了SNN作為聚類的基礎(chǔ),但是前者的總體表現(xiàn)更優(yōu),可能是因為Seurat使用了Louvain算法,而SNN-Cliq則是基于團檢測的方法。
這個實驗表明,即使有大量的專門為單細胞分析所開發(fā)的聚類方法,它們在聚類數(shù)千個細胞時的結(jié)果顯示出相當(dāng)大的變化。并且我們?nèi)匀恍枰恍┓椒ǎ@些方法不像SC3那樣依賴于監(jiān)督學(xué)習(xí),就能夠應(yīng)用于大型數(shù)據(jù)集,例如數(shù)十萬個細胞或者更多。
● 乳腺癌數(shù)據(jù)
我們從公共數(shù)據(jù)中下載得到了來源于11名乳腺癌患者共515個細胞的數(shù)據(jù)集,該數(shù)據(jù)集包含了25636和基因的TPM表達值,我們從中提取了5000個高變基因進行此次分析。這些細胞總體包含三類:免疫細胞、基質(zhì)細胞和腫瘤細胞。由于一些患者的數(shù)據(jù)未覆蓋到全部三種類型,因此,我們最后使用了來自5名患者的212個細胞作為此次實驗的對象。
該數(shù)據(jù)集的主要目的是用于比較兩個適用于混合樣本聚類的工具。首先,Seurat主要通過CCA的方法對來自不同患者的數(shù)據(jù)進行整合。運行Seurat時,我們選取了幾個不同的參數(shù):特征基因數(shù)量分別為{3000,3200,...,5000},典型相關(guān)成分{2,...,10},分辨率{0.2,0.3,0.4,0.5}。通過幾種不同的組合分析,我們最終發(fā)現(xiàn),表現(xiàn)最優(yōu)的組合是1600+2+0.2,分別對應(yīng)上面三個參數(shù)。scVDMC在數(shù)學(xué)優(yōu)化框架中使用內(nèi)嵌的特征選擇來尋找一小組共享的基因以整合數(shù)據(jù)集。我們同樣選取不同的參數(shù)進行組合,最后得到的最優(yōu)組合是λ = 1000,α = 3,w = 3。
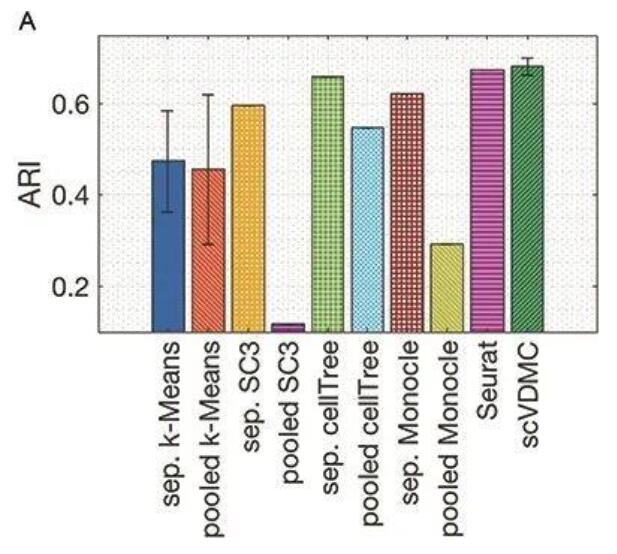
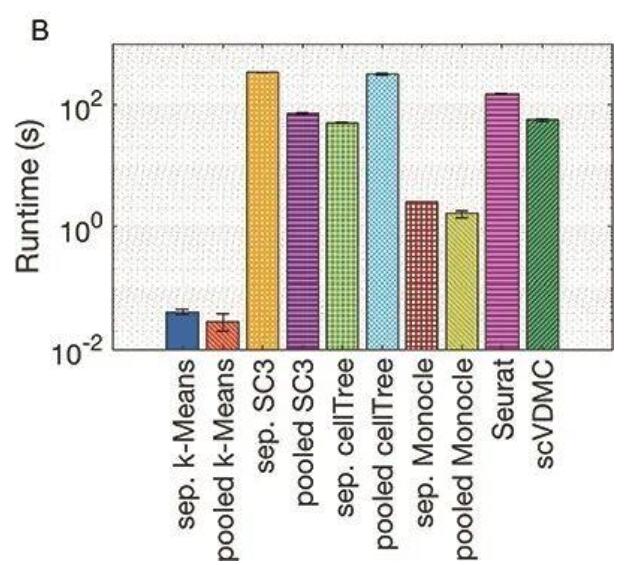
Figure 6 BRCA中聚類方法的比較
(A)Y軸表示ARI值,X軸表示不同工具和數(shù)據(jù)集的組合。(B)同圖A。
我們也將這兩個工具和上一節(jié)中表現(xiàn)最好的Monocle,SC3以及cellTree在兩個層面進行了比較:按照樣本來源分離單獨進行聚類;合并樣本聚類。圖2展示了比較的結(jié)果。從圖6A中我們可以看到,SC3和cellTree在合并聚類的得分中要差很多,提示我們簡單的合并樣本不適用于整合多個不同來源的單細胞數(shù)據(jù)。我們還注意到scVDMC和Seurat都獲得了較高的ARI。其中,scVDMC的平均值為0.681,Seurat的平均值為0.675。盡管scVDMC的均值更高,但其方差也比較大,與Seurat的差異并不具有統(tǒng)計學(xué)意義(p=0.3511)。另外,scVDMC相較于Seurat擁有更少的運行時間(p=2E-14)。總的來說,這些結(jié)果表明對于混合的樣本使用內(nèi)置批次矯正方法(如包含CCA的Seurat)的工具更為有效。
04 討論
在過去的幾年里,專門用于單細胞數(shù)據(jù)分析的聚類算法已經(jīng)有了實質(zhì)性的發(fā)展。這些算法旨在解決單細胞數(shù)據(jù)中固有的挑戰(zhàn),例如細胞特異性偏差、dropouts和技術(shù)噪聲。一些用于解決特定情況(批次、罕見細胞、時間序列)的工具已經(jīng)被開發(fā)出來。此外,不同的方法也越來越關(guān)注數(shù)據(jù)的預(yù)處理,如標(biāo)準化、降維和相似性度量等,這些方法有助于減少執(zhí)行聚類前的技術(shù)差異。總之,這些計算方法的進步為單細胞數(shù)據(jù)的聚類分析提供了非常大的幫助。
我們也注意到,由于單細胞平臺的發(fā)展,細胞捕獲和測序的成本及時間也越來越低,捕獲的細胞數(shù)量越來越高。因此,越來越多的研究更需要擴展性良好的聚類工具或方法,以便能夠在更大的數(shù)據(jù)集中進行使用。而這一發(fā)展也為分析帶來了新的挑戰(zhàn),大多數(shù)現(xiàn)有的工具都無法很好地應(yīng)用到數(shù)萬個甚至更多的單細胞數(shù)據(jù),所以也限制了部分算法在未來研究中的適用性。
另一個現(xiàn)有方法的缺陷是關(guān)于數(shù)據(jù)的整合。如今,單細胞數(shù)據(jù)集仍然在不斷地快速增長,這些開放的大量數(shù)據(jù)將會使我們對特定細胞類型、細胞標(biāo)記、表達模式等擁有更深的了解。此外,這些數(shù)據(jù)還有助于構(gòu)建大規(guī)模不同疾病隊列的單細胞圖譜。然而,目前的聚類方法中,很少有專門應(yīng)用于多數(shù)據(jù)的合并聚類分析,往往需要借助其它工具的使用。
除了本文中主要描述的無監(jiān)督學(xué)習(xí)方法之外,還有一種使用監(jiān)督或半監(jiān)督學(xué)習(xí)方式來進行細胞聚類的替代方法。例如,SC3包使用監(jiān)督學(xué)習(xí)將多余的細胞分配給通過共識聚類發(fā)現(xiàn)的簇,提高了其在大數(shù)據(jù)集應(yīng)用上的延展性。再比如,當(dāng)有一個已知類別的參考數(shù)據(jù)集時,通過Scmap,可以將其他數(shù)據(jù)集中未知的細胞比對到該參考數(shù)據(jù)集中最相似的細胞,從而實現(xiàn)細胞的聚類。
最后,除了單細胞轉(zhuǎn)錄組的數(shù)據(jù)之外,更多不同類型的單細胞組學(xué)方法也呈現(xiàn)風(fēng)靡之勢。盡管面對新類型的單細胞數(shù)據(jù),現(xiàn)有的方法仍然部分適用。但是在未來,也迫切需要應(yīng)對多組學(xué)整合聚類的新的計算方法。
參考文獻
[1] https://blog.bioturing.com/2022/01/27/a-guide-to-scrna-seq-normalization
[2] Campigotto, Romain & Conde-Céspedes, Patricia & Guillaume, Jean-Loup. (2014). A Generalized and Adaptive Method for Community Detection.
[3] Kiselev, V., Kirschner, K., Schaub, M. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 14, 483–486 (2017).
[4] Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019 Jun 13;177(7):1888-1902.e21.

- 翼和生物SNP基因分型技術(shù):揭秘基因密碼,探尋寶寶健康之路
- 促腎上腺皮質(zhì)激素釋放激素(CRH)在探索肥胖機制中的研究
- 泌尿系統(tǒng)疾病的關(guān)鍵治療靶點之V2R的介紹及相關(guān)實驗
- 激光共聚焦顯微鏡助力腫瘤抑制劑研究
- 空間多組學(xué)解碼腫瘤微環(huán)境,助力加速精準治療研究
- 利用單細胞轉(zhuǎn)錄組技術(shù)解析多種免疫細胞對大量細胞因子的響應(yīng)
- Emulate器官芯片助力研究藥物引起肝損傷的機制
- 骨質(zhì)疏松和甲狀旁腺功能減退關(guān)鍵靶點PTH1R的結(jié)構(gòu)功能及相關(guān)實驗介紹