
·ЦЧУ°йӮHҪйҢ§өДЧФКЙҢҰЙсҪӣјҡ°ы·Җ‘BөДЦШТӘХ{№қЧчУГ
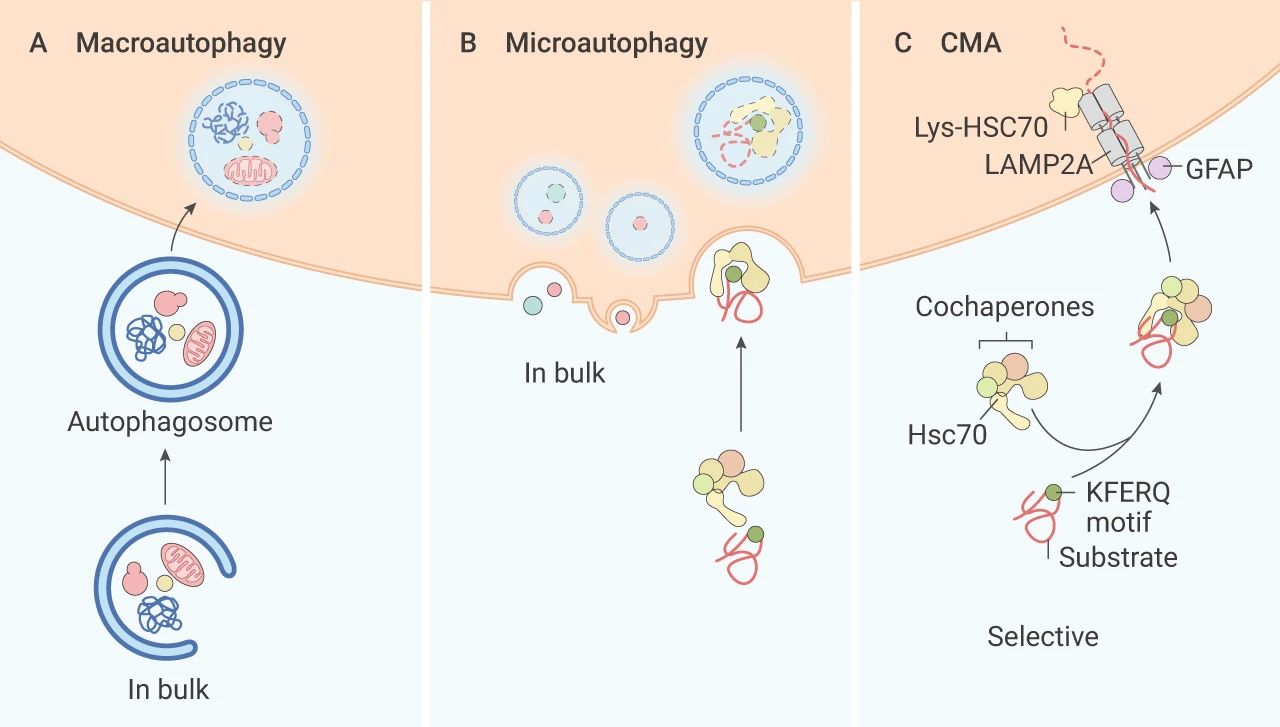
ЧФКЙКЗјҡ°ыФЪНвҪзӯhҫіТтЛШөДУ°н‘ПВЈ¬АыУГИЬГёуwЈ¬ҪөҪвЧФЙнКЬ“pЎўЧғРФҙу·ЦЧУОпЩ|»тХЯјҡ°ыЖчөДЧФОТПы»ҜЯ^іМЎЈТА“юЖд°lЙъНҫҸҪЈ¬ЦчТӘ·ЦһйИэ·NЈәҫЮЧФКЙ (Macroautophagy)Ј¬ОўЧФКЙ (Microautophagy) әН·ЦЧУ°йӮHҪйҢ§өДЧФКЙ (Chaperone-mediated autophagy, CMA)ЎЈ
ҫЮЧФКЙЯ^іМЦРЈ¬ЧФКЙуwөДлpДӨДТЕЭҢўКЬ“pөДө°°ЧЩ|/јҡ°ыЖчӮчЯfЦБИЬГёуwЯMРРҪөҪвЈ¬НЁіЈОД«IЦРМбөҪөДЧФКЙЦёөДКЗҫЮЧФКЙЎЈОўЧФКЙТІНЁЯ^ДТЕЭҢўОпЩ|Э”ЛНөҪИЬГёуwЈ¬ИЬГёуwДӨғИПЭЈ¬ҪөҪв·ҪКҪЦұҪУБЛ®”ЎЈ
·ЦЧУ°йӮHҪйҢ§өДЧФКЙЈәЕcЗ°ғЙ·NЧФКЙІ»Н¬Ј¬°йӮHҪйҢ§өДЧФКЙІ»К№УГДТЕЭЈ¬Я@·NЧФКЙ·ҪКҪҫЯУРёЯ¶ИЯx“сРФЈ¬іЈҪиЦъ°йӮHө°°Ч Hsc70Ј¬МШ®җРФҪөҪвҺ§УРӘҡМШЧR„eОелД»щРт (KFERQ ҳУ) өД°Рө°°ЧЈ¬ИЬГёуwДӨЙПөДКЬуwө°°Ч LAMP2A ЧR„eҪYәПө°°Чұ©В¶өД KFERQ »щҲFЈ¬Ў°ТэҢ§ЎұДҝөДө°°ЧЯMИлИЬГёуwҪөҪвЎЈ
ЙсҪӣНЛРРРФјІІЎЦРЈ¬ЙсҪӣјҡ°ыНщНщ•юАЫ·eҙуБҝҹo·ЁҪөҪвөДө°°ЧЈ¬НЁЯ^ИЬГёуwЧФКЙЯ^іМҪөҪвө°°ЧКЗјҡ°ыЗеіэ®җіЈө°°ЧөДЦчТӘ·ҪКҪЎЈТСУРҲуөАЈ¬CMA Йжј°ЙсҪӣНЛРРРФјІІЎІЎФӯө°°ЧЩ| (Из ҰБ-syn әН tau) өДҪөҪвЈ¬И»¶шкPУЪЙсҪӣНЛРРРФјІІЎЦР CMA өДК§ИҘ№ҰДЬҢ§ЦВөДәу№ыЙРОҙЗеіюЎЈ
ҪьИХЈ¬Ana Maria Cuervo өИИЛФЪ Cell ЙП°lұнБЛо}һй Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome өДОДХВЈ¬ҪТКҫБЛ CMA ҢҰЙсҪӣјҡ°ы·Җ‘BөДЦШТӘХ{№қЧчУГЎЈ

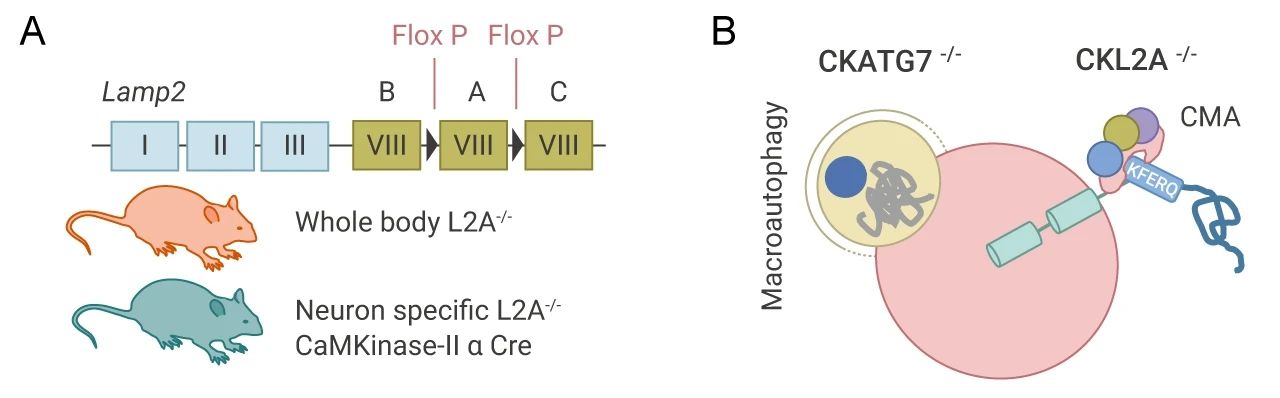
ФЪЯ@ЖӘОДХВЦРЈ¬СРҫҝХЯӮғ·Ц„eҳӢҪЁБЛИ«ЙнРФ LAMP2A (L2A) ЗГіэДЈРН (L2A-/-) әНЙсҪӣФӘ—lјюЗГіэ L2A/LAMP2a (CKL2A-/-) РЎКуДЈРН (ҲD 3A)Ј¬Ғн·ЦОц CMA ҢҰҫSіЦЙсҪӣФӘө°°ЧЩ|·Җ‘BөДЧчУГЎЈЯҖҪЁБўБЛДЈ”MҫЮЧФКЙИұК§ (ATG7 өД—lјюРФЗГіэ) CKATG7-/- РЎКуДЈРН (ҲD 3B)Ј¬ІўЕc CKL2A-/- РЎКуДЈРНҢҰұИЈ¬Ғн·ЦОц CMA әНҫЮЧФКЙЯ@ғЙ—lЧФКЙНЁВ·ҢҰЙсҪӣФӘө°°ЧЧғРФөДМШ¶ЁЧчУГЎЈ(CTR ҪMРЎКуһйІ»ЧцЗГіэөДҢҰХХҪM)ЎЈ
Ўц CKL2A-/- РЎКуәН L2A-/- РЎКуЛщұн¬FіцөДЙсҪӣ“pәҰРРһй
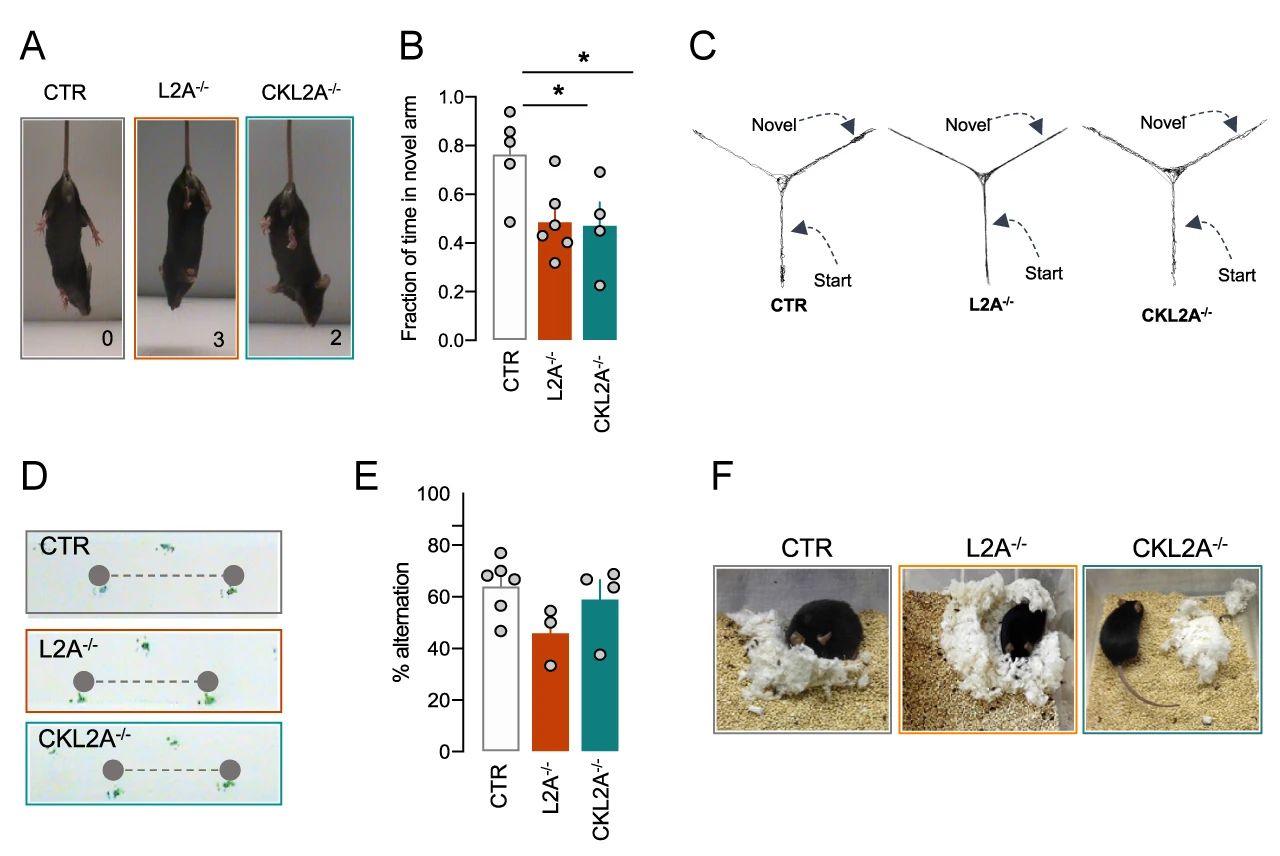
КЧПИЈ¬СРҫҝХЯӮғ°l¬FЈ¬CKL2A-/- РЎКуәН L2A-/- РЎКуЕcҢҰХХҪM (CTR) ПаұИЈ¬РРһйҢWңyФҮөГ·ЦёьёЯЈ¬әуЦ«ҫoОХЯMХ№ёьҝм (ҲD 4A)Ј¬Я@ғЙ·NРЎКуЯҖҫщұн¬FіцБЛёРУXЯ\„У№ҰДЬХПөKәН Y РНГФҢmЦР¶М•rУӣ‘ӣңpЙЩ (ҲD 4B-C) ЎЈЦ»УР L2A-/- РЎКуұн¬FіцоҗЛЖЕБҪрЙӯјІІЎөДІҪ‘BМШХчЈ¬ИзІҪ·щңpЙЩ (ҲD 4D)ЎЈҙЛНвЈ¬CKL2A-/- РЎКуЯҖұн¬FіцБЛЙсҪӣЧғРФөДіЈТҠұнРНЈ¬ИзУРҝХйg№ӨЧчУӣ‘ӣңpЙЩәНЦюіІРРһйп@ЦшңpЙЩөДЪ…„Э (ҲD 4E-F)ЎЈ
ТФЙПҪY№ып@КҫЈ¬ЙсҪӣФӘ L2A ИұК§өДРЎКуұн¬Fіц L2A ПөҪyРФИұК§РЎКуөДҙуІҝ·ЦРРһйЭ”іцЎЈ
Ўц ЙсҪӣФӘ CMA ИұПЭҢ§ЦВө°°ЧЩ|·Җ‘BөДЖЖүД
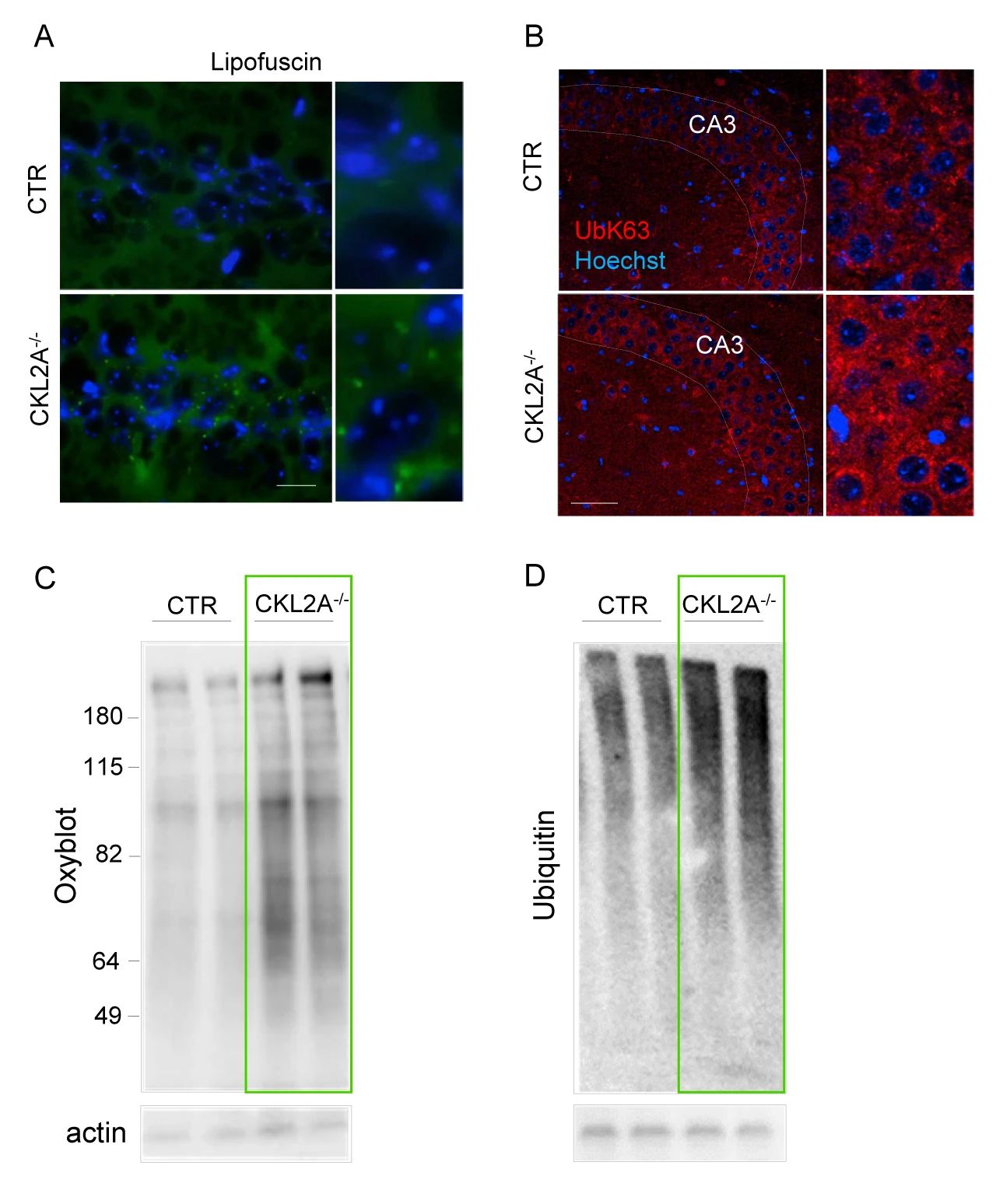
ЧчХЯҲFк °l¬FЈ¬ФЪ 6 ӮҖФВҙуөД L2A-/- РЎКуөДәЈсRуwЦРЈ¬іц¬FБЛЦ¬әЦЩ|әН K63 ·әЛШ»Ҝө°°ЧөД·eАЫ (ғЙӮҖө°°ЧКЗИЬГёуwҪөҪвөД°Рө°°Ч)Ј¬CKL2A-/- РЎКуөДәЈсRуwөДеFуwЙсҪӣФӘЦРТІ°l¬FБЛоҗЛЖМШХч (ҲD 5A-B)ЎЈН¬•r CKL2A-/- РЎКуЖӨЩ|өДГвТЯУЎЫE·ЦОцп@КҫЈ¬Сх»Ҝө°°Ч·әЛШ»Ҝө°°ЧАЫ·e (ҲD 5C-D)Ј¬Я@Р©ҪY№ыұнГч CMA ИұК§•юҢ§ЦВЙсҪӣФӘө°°Ч·Җ‘BөДЖЖүДЎЈ
ТФЙПҪY№ыұнГчЈ¬ФЪ CMA ИұПЭ•rЮDЧғһйІ»ИЬРФөДө°°ЧЩ|ҪMКЗЯ^п–әНө°°ЧЩ|ҪMЦРөДТ»Іҝ·ЦЈ¬ІўЗТЙсҪӣФӘ CMA ИұК§•юҢ§ЦВө°°ЧЩ|ҪMөДЖЖүДЎЈ
Ўц CMA әНҫЮЧФКЙҢҰЙсҪӣФӘө°°ЧЩ|ЧғРФөДУ°н‘І»Н¬
ПВТ»ІҪЈ¬СРҫҝХЯӮғФЪ CKATG7-/- РЎКуәН CKL2A-/- РЎКуДЈРНЦРЈ¬НЁЯ^ұИЭ^ө°°ЧЩ|ҪMҢWҒнкUГч CMA әНҫЮЧФКЙҢҰЙсҪӣФӘө°°ЧЩ|ЧғРФөДМШ¶ЁЧчУГЎЈ
І»ИЬРФө°°ЧЩ|ҪMөД»щТтјҜё»јҜәНё»јҜҲD·ЦОц (ҲD 7A) ҪY№ыұнГчЈ¬CKATG7-/- РЎКуәН CKL2A-/- РЎКуЦР°lЙъЧғ»ҜөДө°°ЧЩ|ҪMІ»ПаН¬ЎЈCKATG7-/- Ң§ЦВјҡ°ыЦЬЖЪәН·әЛШ»Ҝө°°ЧГёуw·ЦҪвҙъЦxЯ^іМПакPөДө°°ЧЧғ»Ҝ (Л{Й«јэо^Цёіц)Ј¬¶шЕc CMA ИұПЭЕcө°°ЧЩ|Я\Э”әНҙъЦxУРкP (јtЙ«јэо^Цёіц)ЎЈ
јҡ°ыНвЛб»ҜВК (ECAR) өДңy¶ЁҪY№ыұнГчЈ¬CKL2A-/- ЙсҪӣФӘЦРМЗҪНҪвөДп@ЦшңpЙЩ (ҲD 7B)Ј¬¶шЗТәЬ¶аМЗҪНҪвГёТІФЪ CKL2A-/- РЎКуөДІ»ИЬРФө°°ЧІҝ·ЦЦРФцјУЈ¬ИзұыНӘЛбГ“ҡдГё (PDH) (ҲD 7C)ЎЈ
лmИ»ТСУРҲуөАЧи”аҫЮЧФКЙТІ•юУ°н‘ЙсҪӣФӘМЗҪНҪвЈ¬ө«ҢҚтһҪY№ып@КҫЈ¬L2A әН ATG7 ЗГөНҢҰМЗҪНҪвМШРФөДУ°н‘ІўІ»Н¬ЎЈТФЙПҪY№ыұнГчЈ¬CMA ИұПЭЙсҪӣФӘө°°ЧЩ|ҪMПтІ»ИЬРФө°°ЧЮDЧғәуҢ§ЦВөДҪY№ы…^„eУЪҫЮЧФКЙЎЈ
»щУЪЙПГжөДСРҫҝЈ¬НЖ”аЈәЙсҪӣФӘ CMA ұ»ЦұҪУЧи”аәуЈ¬І»ИЬРФө°°ЧөД·eАЫәНЙсҪӣФӘ№ҰДЬөДёДЧғЈ¬Я@ҝЙДЬФцјУЙсҪӣНЛРРРФјІІЎөДТЧ“pРФІўјУЛЩјІІЎЯMХ№ЎЈ
СРҫҝХЯҪЁБўБЛ hTauP301L Я^ұнЯ_РЎКуЈ¬ҪY№ыұнГчЈәЖдЙсҪӣјҡ°ыөД CMA »оРФҪөөНЈ¬CMA °Яьc”өДҝГчп@ңpЙЩ (ҲD 8A)ЎЈСРҫҝХЯЯҖҪЁБўБЛ P301S tau ЮD»щТтРЎКуЈ¬ФЪФ“ДЈРНЦРК№УГБЛ CA77.1 (AR7 өДСЬЙъОп) ФЪуwНвјӨ»о CMA ЗТІ»У°н‘ҫЮЧФКЙ (ҲD 8B)ЎЈФЪуwғИЈ¬CA77.1 ҪoЛҺК№өГФӯұҫЎ°БиҒyЎұөД PS19 РЎКуөДХэіЈ»Ҝ (ҲD 8C)Ј¬Іўп@ЦшңpЙЩБЛәЈсRЎўРУИКәЛәНАж оЖӨҢУЦРә¬УРЦВІЎРФ tau ҳӢПуөДЙсҪӣФӘөД”өБҝ (ҲD 8D)ЎЈ
ҝӮҪYЈә
Я@ЖӘОДХВід·ЦЧCГчБЛ·ЦЧУ°йӮHЧФКЙ (CMA) ҢҰЙсҪӣјҡ°ы·Җ‘BҫЯУРЦШТӘХ{№қЧчУГЎЈ
СРҫҝХЯӮғНЁЯ^ҫЯУРИ«ЙнРФәНЙсҪӣФӘМШ®җРФ CMA Чи”аөДРЎКуДЈРНЈ¬ЧCГчБЛЙсҪӣФӘ CMA өДИұК§Ң§ЦВө°°ЧЩ|¶ҫРФәНЙсҪӣФӘ№ҰДЬХПөKЎЈCMA ИұК§ЯҖ•юК№ФӯұҫТЧУЪҫЫјҜөД KFERQ ҳУ»щРтө°°ЧЩ|ПтІ»ИЬРФө°°ЧЮDЧғЎЈ
НЁЯ^ЙсҪӣФӘМШ®җРФ CMA әН ATG7 ИұК§РЎКуДЈРНЈ¬ЧCГчБЛ CMA әНҫЮЧФКЙФЪХ{ҝШЙсҪӣФӘө°°ЧЩ|·Җ‘BЦРЕcЙсҪӣЧғРФөДҒҶө°°ЧЩ|ҪMІўІ»ПаН¬ЎЈ
MCE өДЛщУР®aЖ·ғHУГЧчҝЖҢWСРҫҝ»тЛҺЧCЙкҲуЈ¬ОТӮғІ»һйИОәОӮҖИЛУГНҫМṩ®aЖ·әН·ю„Х
…ўҝјОД«I
1. Evripidis Gavathiotis, Ana Maria Cuerv, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell2021 May 13;184(10):2696-2714.e25.
2. Susmita Kaushik, Ana Maria Cuervo, et al. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):365-381.
3. Esteban-Martı´nez, L., Sierra-Filardi, E., McGreal,R.S., Salazar-Roa, M, et al. Programmed mitophagy is essential for theglycolytic switch during cell differentiation.EMBO J. 36, 1688-1706.
ҫЮЧФКЙЯ^іМЦРЈ¬ЧФКЙуwөДлpДӨДТЕЭҢўКЬ“pөДө°°ЧЩ|/јҡ°ыЖчӮчЯfЦБИЬГёуwЯMРРҪөҪвЈ¬НЁіЈОД«IЦРМбөҪөДЧФКЙЦёөДКЗҫЮЧФКЙЎЈОўЧФКЙТІНЁЯ^ДТЕЭҢўОпЩ|Э”ЛНөҪИЬГёуwЈ¬ИЬГёуwДӨғИПЭЈ¬ҪөҪв·ҪКҪЦұҪУБЛ®”ЎЈ
·ЦЧУ°йӮHҪйҢ§өДЧФКЙЈәЕcЗ°ғЙ·NЧФКЙІ»Н¬Ј¬°йӮHҪйҢ§өДЧФКЙІ»К№УГДТЕЭЈ¬Я@·NЧФКЙ·ҪКҪҫЯУРёЯ¶ИЯx“сРФЈ¬іЈҪиЦъ°йӮHө°°Ч Hsc70Ј¬МШ®җРФҪөҪвҺ§УРӘҡМШЧR„eОелД»щРт (KFERQ ҳУ) өД°Рө°°ЧЈ¬ИЬГёуwДӨЙПөДКЬуwө°°Ч LAMP2A ЧR„eҪYәПө°°Чұ©В¶өД KFERQ »щҲFЈ¬Ў°ТэҢ§ЎұДҝөДө°°ЧЯMИлИЬГёуwҪөҪвЎЈ
ҲD 1. Иэ·NІ»Н¬өДЧФКЙНҫҸҪ[2]
A. ҫЮЧФКЙЈ»B. ОўЧФКЙЈ»C. °йӮHҪйҢ§өДЧФКЙ
A. ҫЮЧФКЙЈ»B. ОўЧФКЙЈ»C. °йӮHҪйҢ§өДЧФКЙ
ЙсҪӣНЛРРРФјІІЎЦРЈ¬ЙсҪӣјҡ°ыНщНщ•юАЫ·eҙуБҝҹo·ЁҪөҪвөДө°°ЧЈ¬НЁЯ^ИЬГёуwЧФКЙЯ^іМҪөҪвө°°ЧКЗјҡ°ыЗеіэ®җіЈө°°ЧөДЦчТӘ·ҪКҪЎЈТСУРҲуөАЈ¬CMA Йжј°ЙсҪӣНЛРРРФјІІЎІЎФӯө°°ЧЩ| (Из ҰБ-syn әН tau) өДҪөҪвЈ¬И»¶шкPУЪЙсҪӣНЛРРРФјІІЎЦР CMA өДК§ИҘ№ҰДЬҢ§ЦВөДәу№ыЙРОҙЗеіюЎЈ
ҪьИХЈ¬Ana Maria Cuervo өИИЛФЪ Cell ЙП°lұнБЛо}һй Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome өДОДХВЈ¬ҪТКҫБЛ CMA ҢҰЙсҪӣјҡ°ы·Җ‘BөДЦШТӘХ{№қЧчУГЎЈ
ҲD 2. Ana Maria Cuervo өИИЛ°lұнөД CMA ПакPОДХВ
ФЪЯ@ЖӘОДХВЦРЈ¬СРҫҝХЯӮғ·Ц„eҳӢҪЁБЛИ«ЙнРФ LAMP2A (L2A) ЗГіэДЈРН (L2A-/-) әНЙсҪӣФӘ—lјюЗГіэ L2A/LAMP2a (CKL2A-/-) РЎКуДЈРН (ҲD 3A)Ј¬Ғн·ЦОц CMA ҢҰҫSіЦЙсҪӣФӘө°°ЧЩ|·Җ‘BөДЧчУГЎЈЯҖҪЁБўБЛДЈ”MҫЮЧФКЙИұК§ (ATG7 өД—lјюРФЗГіэ) CKATG7-/- РЎКуДЈРН (ҲD 3B)Ј¬ІўЕc CKL2A-/- РЎКуДЈРНҢҰұИЈ¬Ғн·ЦОц CMA әНҫЮЧФКЙЯ@ғЙ—lЧФКЙНЁВ·ҢҰЙсҪӣФӘө°°ЧЧғРФөДМШ¶ЁЧчУГЎЈ(CTR ҪMРЎКуһйІ»ЧцЗГіэөДҢҰХХҪM)ЎЈ
ҲD 3. ҪЁБўРЎКуҢҚтһДЈРН
A. L2A өДИ«ЙнРФЗГіэәН—lјюЗГіэЈ»B. ATG7 өД—lјюРФЗГіэ
A. L2A өДИ«ЙнРФЗГіэәН—lјюЗГіэЈ»B. ATG7 өД—lјюРФЗГіэ
Ўц CKL2A-/- РЎКуәН L2A-/- РЎКуЛщұн¬FіцөДЙсҪӣ“pәҰРРһй
КЧПИЈ¬СРҫҝХЯӮғ°l¬FЈ¬CKL2A-/- РЎКуәН L2A-/- РЎКуЕcҢҰХХҪM (CTR) ПаұИЈ¬РРһйҢWңyФҮөГ·ЦёьёЯЈ¬әуЦ«ҫoОХЯMХ№ёьҝм (ҲD 4A)Ј¬Я@ғЙ·NРЎКуЯҖҫщұн¬FіцБЛёРУXЯ\„У№ҰДЬХПөKәН Y РНГФҢmЦР¶М•rУӣ‘ӣңpЙЩ (ҲD 4B-C) ЎЈЦ»УР L2A-/- РЎКуұн¬FіцоҗЛЖЕБҪрЙӯјІІЎөДІҪ‘BМШХчЈ¬ИзІҪ·щңpЙЩ (ҲD 4D)ЎЈҙЛНвЈ¬CKL2A-/- РЎКуЯҖұн¬FіцБЛЙсҪӣЧғРФөДіЈТҠұнРНЈ¬ИзУРҝХйg№ӨЧчУӣ‘ӣңpЙЩәНЦюіІРРһйп@ЦшңpЙЩөДЪ…„Э (ҲD 4E-F)ЎЈ
ТФЙПҪY№ып@КҫЈ¬ЙсҪӣФӘ L2A ИұК§өДРЎКуұн¬Fіц L2A ПөҪyРФИұК§РЎКуөДҙуІҝ·ЦРРһйЭ”іцЎЈ
ҲD 4. L2A ИұК§РЎКуұн¬FіцРРһйХПөKA. КуЧҰЧҘБҰңy¶ЁЈ»B-CЎўE. Y РНГФҢmРРһйҢWҷzңyЈ»D. ІҪ‘B·ЦОцЈ»F. ЦюіІФҮтһ
Ўц ЙсҪӣФӘ CMA ИұПЭҢ§ЦВө°°ЧЩ|·Җ‘BөДЖЖүД
ЧчХЯҲFк °l¬FЈ¬ФЪ 6 ӮҖФВҙуөД L2A-/- РЎКуөДәЈсRуwЦРЈ¬іц¬FБЛЦ¬әЦЩ|әН K63 ·әЛШ»Ҝө°°ЧөД·eАЫ (ғЙӮҖө°°ЧКЗИЬГёуwҪөҪвөД°Рө°°Ч)Ј¬CKL2A-/- РЎКуөДәЈсRуwөДеFуwЙсҪӣФӘЦРТІ°l¬FБЛоҗЛЖМШХч (ҲD 5A-B)ЎЈН¬•r CKL2A-/- РЎКуЖӨЩ|өДГвТЯУЎЫE·ЦОцп@КҫЈ¬Сх»Ҝө°°Ч·әЛШ»Ҝө°°ЧАЫ·e (ҲD 5C-D)Ј¬Я@Р©ҪY№ыұнГч CMA ИұК§•юҢ§ЦВЙсҪӣФӘө°°Ч·Җ‘BөДЖЖүДЎЈ
ҲD 5. CMA ИұК§•юҢ§ЦВЙсҪӣФӘө°°Ч·Җ‘BөДЖЖүДA. әЈсRуwЦРөДЦ¬әЦЛШҹЙ№вҷzңyЈ»B. K63 ·әЛШ»Ҝө°°ЧөДҷzңyЈ»C-D. Сх»Ҝө°°ЧәН·әЛШ»Ҝө°°Ч·eАЫөДҷzңy
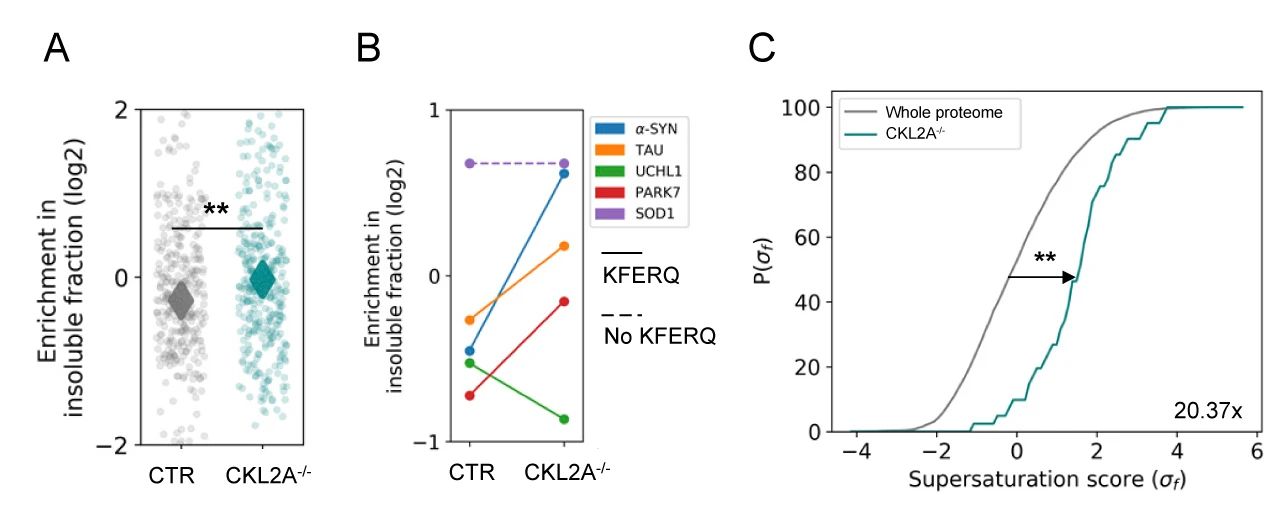
ҫoҪУЦшЈ¬СРҫҝХЯӮғ·Цлx CTR ҪMәН CKL2A-/- РЎКуЖӨҢУ Sarkosy І»ҝЙИЬө°°ЧЈ¬ЯMРРБЛ¶ЁБҝө°°ЧҪMҢWөД·ЦОцЎЈҢҚтһҪY№ыұнГч CKL2A-/- РЎКуҙуДXҫЫјҜБЛҙуБҝІ»ҝЙИЬө°°ЧЈ¬ЗТЯ@Р©ө°°Ч 76ЈҘ ә¬УР KFERQ ҳУ»щРтЈ¬ФӯұҫТЧУЪҫЫјҜөДҺ§УР KFERQ ҳУ»щРтөДө°°ЧЩ| (АэИз ҰБ-synЎўtauЎўUCHL1 әН PARK7 өИө°°Ч) ПтІ»ИЬРФө°°ЧөДЮDЧғФц¶а (ҲD 6B)ЎЈСРҫҝХЯӮғЯҖ·ЦОцБЛЯ^п–әН·Ц”өЈ¬°l¬F CKL2A-/- РЎКуөДІ»ИЬРФө°°ЧЩ|ЦРөДЯ^п–әН·Ц”өп@ЦшФцјУ (20.37 ұ¶)ЎЈ
ҲD 6. ЙсҪӣФӘ CMA ұ»Чи”аәуө°°ЧЩ|ҪMөДЧғ»ҜA. ҝӮө°°ЧЩ|әНТЧУЪҫЫјҜөДө°°ЧЩ|өДё»јҜЈ»B. Һ§УР KFERQ ҳУ»щРтөДө°°ЧЩ|ПтІ»ИЬРФө°°ЧЮDЧғЈ»C. Я^п–әНө°°Ч·Ц”өөДФцјУ
ТФЙПҪY№ыұнГчЈ¬ФЪ CMA ИұПЭ•rЮDЧғһйІ»ИЬРФөДө°°ЧЩ|ҪMКЗЯ^п–әНө°°ЧЩ|ҪMЦРөДТ»Іҝ·ЦЈ¬ІўЗТЙсҪӣФӘ CMA ИұК§•юҢ§ЦВө°°ЧЩ|ҪMөДЖЖүДЎЈ
Ўц CMA әНҫЮЧФКЙҢҰЙсҪӣФӘө°°ЧЩ|ЧғРФөДУ°н‘І»Н¬
ПВТ»ІҪЈ¬СРҫҝХЯӮғФЪ CKATG7-/- РЎКуәН CKL2A-/- РЎКуДЈРНЦРЈ¬НЁЯ^ұИЭ^ө°°ЧЩ|ҪMҢWҒнкUГч CMA әНҫЮЧФКЙҢҰЙсҪӣФӘө°°ЧЩ|ЧғРФөДМШ¶ЁЧчУГЎЈ
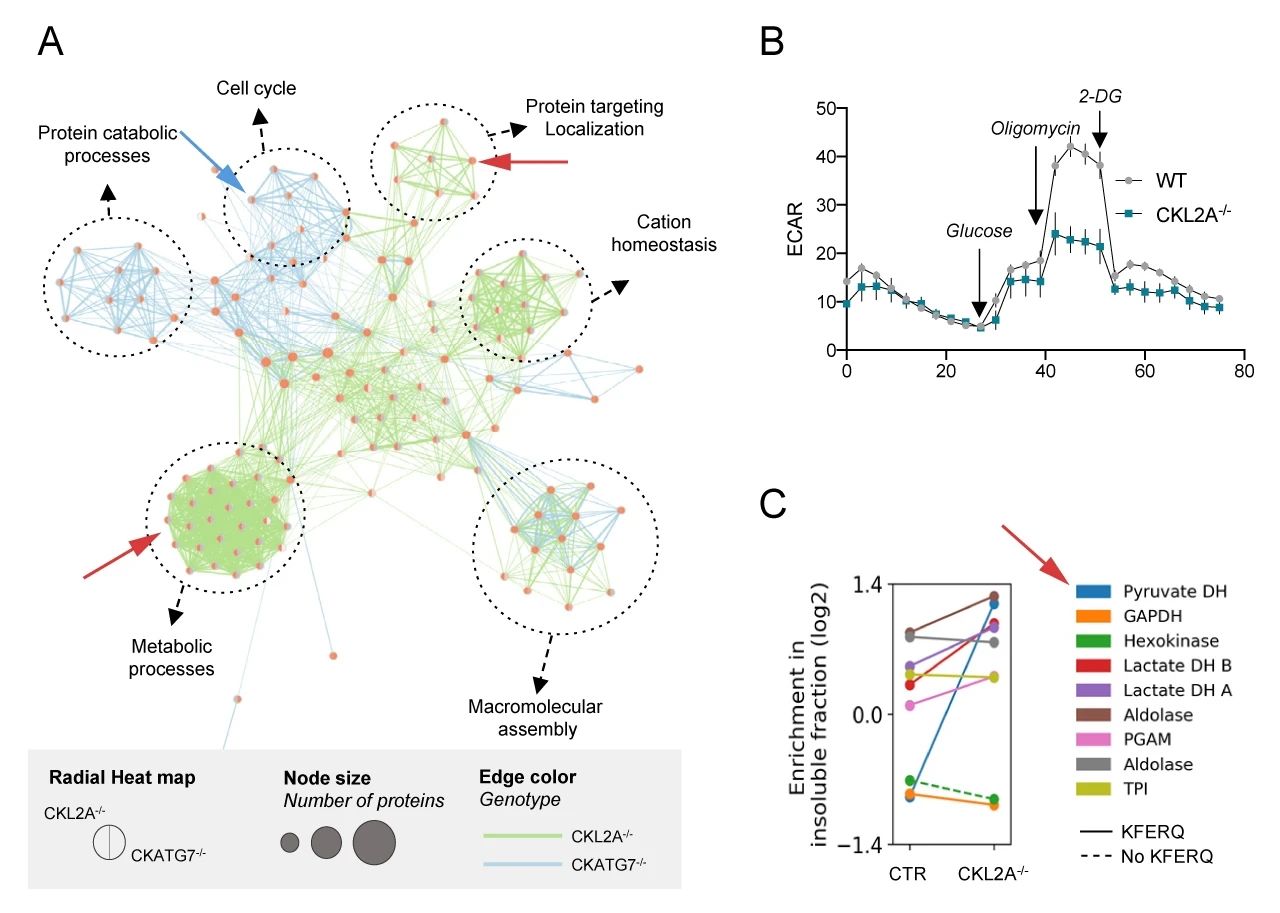
І»ИЬРФө°°ЧЩ|ҪMөД»щТтјҜё»јҜәНё»јҜҲD·ЦОц (ҲD 7A) ҪY№ыұнГчЈ¬CKATG7-/- РЎКуәН CKL2A-/- РЎКуЦР°lЙъЧғ»ҜөДө°°ЧЩ|ҪMІ»ПаН¬ЎЈCKATG7-/- Ң§ЦВјҡ°ыЦЬЖЪәН·әЛШ»Ҝө°°ЧГёуw·ЦҪвҙъЦxЯ^іМПакPөДө°°ЧЧғ»Ҝ (Л{Й«јэо^Цёіц)Ј¬¶шЕc CMA ИұПЭЕcө°°ЧЩ|Я\Э”әНҙъЦxУРкP (јtЙ«јэо^Цёіц)ЎЈ
јҡ°ыНвЛб»ҜВК (ECAR) өДңy¶ЁҪY№ыұнГчЈ¬CKL2A-/- ЙсҪӣФӘЦРМЗҪНҪвөДп@ЦшңpЙЩ (ҲD 7B)Ј¬¶шЗТәЬ¶аМЗҪНҪвГёТІФЪ CKL2A-/- РЎКуөДІ»ИЬРФө°°ЧІҝ·ЦЦРФцјУЈ¬ИзұыНӘЛбГ“ҡдГё (PDH) (ҲD 7C)ЎЈ
ҲD 7. ЙсҪӣФӘө°°ЧЩ|ҪMІ»Н¬ҒҶИәөДЧғ»Ҝ·ЦОц
A. І»ИЬРФө°°Ч»щТтјҜё»јҜ·ЦОцЈ»B. јҡ°ыНвЛб»ҜВК (ECAR) өДңy¶ЁЈ»C: МЗҪНҪвГёұнЯ_өДңpЙЩ
A. І»ИЬРФө°°Ч»щТтјҜё»јҜ·ЦОцЈ»B. јҡ°ыНвЛб»ҜВК (ECAR) өДңy¶ЁЈ»C: МЗҪНҪвГёұнЯ_өДңpЙЩ
лmИ»ТСУРҲуөАЧи”аҫЮЧФКЙТІ•юУ°н‘ЙсҪӣФӘМЗҪНҪвЈ¬ө«ҢҚтһҪY№ып@КҫЈ¬L2A әН ATG7 ЗГөНҢҰМЗҪНҪвМШРФөДУ°н‘ІўІ»Н¬ЎЈТФЙПҪY№ыұнГчЈ¬CMA ИұПЭЙсҪӣФӘө°°ЧЩ|ҪMПтІ»ИЬРФө°°ЧЮDЧғәуҢ§ЦВөДҪY№ы…^„eУЪҫЮЧФКЙЎЈ
»щУЪЙПГжөДСРҫҝЈ¬НЖ”аЈәЙсҪӣФӘ CMA ұ»ЦұҪУЧи”аәуЈ¬І»ИЬРФө°°ЧөД·eАЫәНЙсҪӣФӘ№ҰДЬөДёДЧғЈ¬Я@ҝЙДЬФцјУЙсҪӣНЛРРРФјІІЎөДТЧ“pРФІўјУЛЩјІІЎЯMХ№ЎЈ
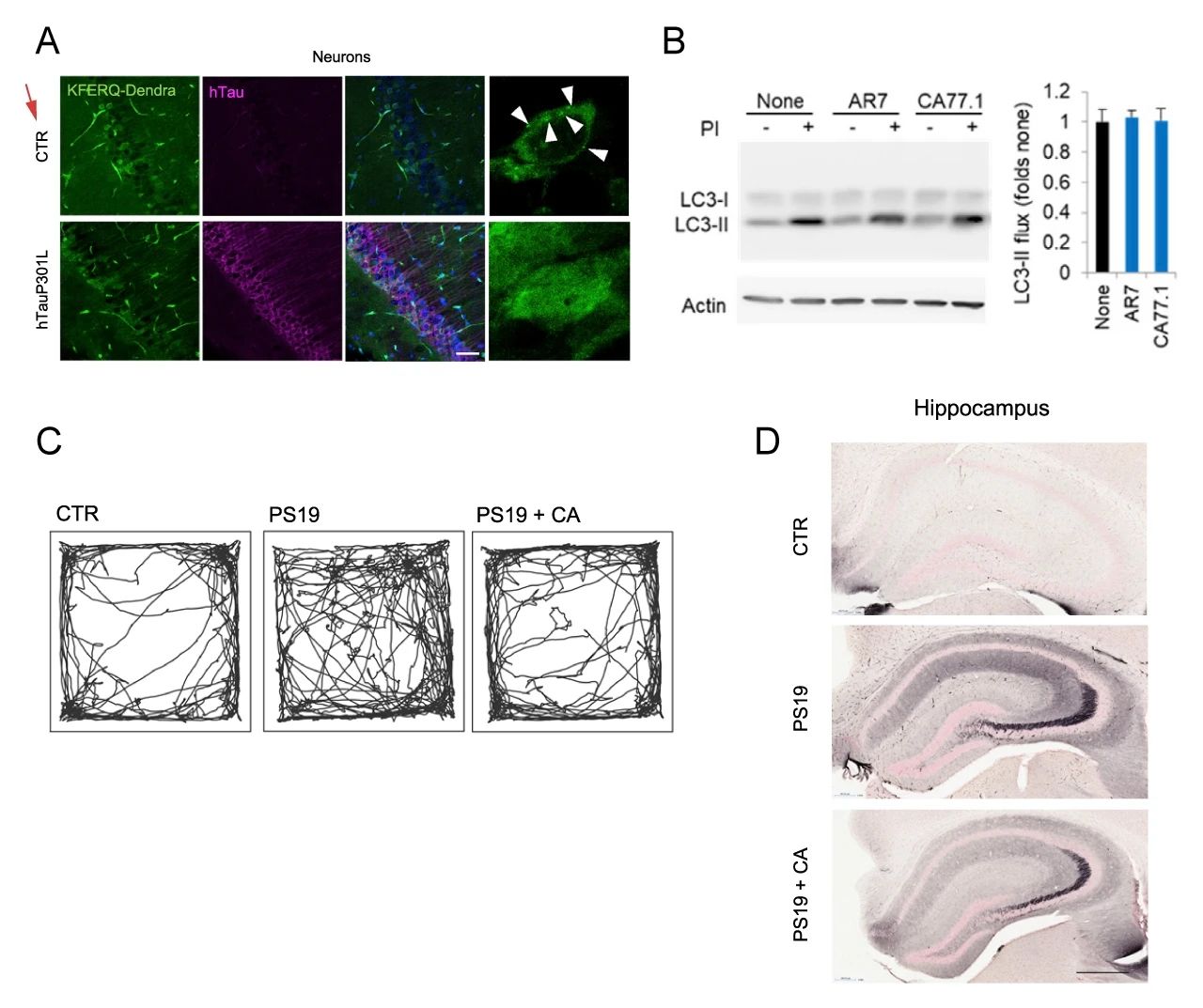
СРҫҝХЯҪЁБўБЛ hTauP301L Я^ұнЯ_РЎКуЈ¬ҪY№ыұнГчЈәЖдЙсҪӣјҡ°ыөД CMA »оРФҪөөНЈ¬CMA °Яьc”өДҝГчп@ңpЙЩ (ҲD 8A)ЎЈСРҫҝХЯЯҖҪЁБўБЛ P301S tau ЮD»щТтРЎКуЈ¬ФЪФ“ДЈРНЦРК№УГБЛ CA77.1 (AR7 өДСЬЙъОп) ФЪуwНвјӨ»о CMA ЗТІ»У°н‘ҫЮЧФКЙ (ҲD 8B)ЎЈФЪуwғИЈ¬CA77.1 ҪoЛҺК№өГФӯұҫЎ°БиҒyЎұөД PS19 РЎКуөДХэіЈ»Ҝ (ҲD 8C)Ј¬Іўп@ЦшңpЙЩБЛәЈсRЎўРУИКәЛәНАж оЖӨҢУЦРә¬УРЦВІЎРФ tau ҳӢПуөДЙсҪӣФӘөД”өБҝ (ҲD 8D)ЎЈ
ҲD 8. CMA өД»ҜҢWјӨ»оёДЙЖБЛ hTauP301L әН PS19 РЎКуөДЙсҪӣІЎАнМШХч
A. hTauP301L РЎКуЙсҪӣФӘ CMA ИҫЙ«Ј»B. CA77.1 уwНвјӨ»о CMAЈ»C. P301S tau РЎКуөДРРһйңyФҮЈ»D. әЈсRуwГвТЯҪM»ҜИҫЙ«
A. hTauP301L РЎКуЙсҪӣФӘ CMA ИҫЙ«Ј»B. CA77.1 уwНвјӨ»о CMAЈ»C. P301S tau РЎКуөДРРһйңyФҮЈ»D. әЈсRуwГвТЯҪM»ҜИҫЙ«
ҝӮҪYЈә
Я@ЖӘОДХВід·ЦЧCГчБЛ·ЦЧУ°йӮHЧФКЙ (CMA) ҢҰЙсҪӣјҡ°ы·Җ‘BҫЯУРЦШТӘХ{№қЧчУГЎЈ
СРҫҝХЯӮғНЁЯ^ҫЯУРИ«ЙнРФәНЙсҪӣФӘМШ®җРФ CMA Чи”аөДРЎКуДЈРНЈ¬ЧCГчБЛЙсҪӣФӘ CMA өДИұК§Ң§ЦВө°°ЧЩ|¶ҫРФәНЙсҪӣФӘ№ҰДЬХПөKЎЈCMA ИұК§ЯҖ•юК№ФӯұҫТЧУЪҫЫјҜөД KFERQ ҳУ»щРтө°°ЧЩ|ПтІ»ИЬРФө°°ЧЮDЧғЎЈ
НЁЯ^ЙсҪӣФӘМШ®җРФ CMA әН ATG7 ИұК§РЎКуДЈРНЈ¬ЧCГчБЛ CMA әНҫЮЧФКЙФЪХ{ҝШЙсҪӣФӘө°°ЧЩ|·Җ‘BЦРЕcЙсҪӣЧғРФөДҒҶө°°ЧЩ|ҪMІўІ»ПаН¬ЎЈ
| ·ЦЧУ°йӮHҪйҢ§өДЧФКЙПакP®aЖ· |
| AR7 уwНвМШ®җРФјӨ»о°йӮHҪйҢ§өДЧФКЙ (CMA) »оРФЈ¬ЗТІ»У°н‘ҫЮЧФКЙЎЈ |
| QX77 °йӮHҪйҢ§ЧФКЙ (CMA) јӨ»о„©Ј¬AR7 өДҪYҳӢСЬЙъОпЎЈQX77 ХTҢ§ Rab11ұнЯ_ЙПХ{ЎЈ |
| CA77.1 AR7 өДСЬЙъОпЈ¬ҫЯУРДXқBНёРФәНҝЪ·ю»оРФөД CMA өДјӨ»о„©Ј¬ҝЙУГУЪЙсҪӣПакPјІІЎөДСРҫҝЎЈ |
| Thioflavine S ҹЙ№вҳЛУӣОпЈ¬ҝЙИҫЙ«өн·ЫҳУ°ЯүKЈ¬ҝЙУГУЪ°ў –ЖқәЈД¬°YөИјІІЎөДСРҫҝЎЈ |
| Hoechst 33342 °lЛ{Й«ҹЙ№вөД DNA ИҫБПЈ¬ҝЙНЁНёЯ^јҡ°ыДӨЎЈ |
| ҰВ-Amyloid ө°°ЧПакP®aЖ· |
| ҰВ-Amyloid (1-42), human TFA ҰВ-Amyloid (1-42), human TFA КЗУЙ 42 ӮҖ°ұ»щЛбҪMіЙөДлДЈ¬КЗҳӢіЙАПДк°ЯәНЙсҪӣАwҫSАpҪYөДЦчТӘіЙ·ЦЈ¬ФЪ°ў –ҙДәЈД¬ІЎөД°lІЎҷCЦЖЦРЖркPжIЧчУГЎЈ |
| ҰВ-Amyloid (25-35) ҰВ-Amyloid (25-35) КЗ ҰВ-өн·ЫҳУө°°ЧөД»оРФЖ¬¶ОЈ¬ҝЙХTҢ§°ў –ҙДәЈД¬ІЎПакPөДЙсҪӣ¶ҫРФЎЈ |
| ҰВ-Amyloid (1-42), rat TFA ҰВ-Amyloid (1-42), rat TFA КЗУЙ 42 ӮҖ°ұ»щЛбҪMіЙөДлДЈ¬УРЙсҪӣ¶ҫРФЧчУГЈ¬ҝЙУГУЪ°ў –ЖқәЈД¬°YөДПакPСРҫҝЎЈ |
| ҰВ-Amyloid (42-1), human Amyloid ҰВ Peptide (42-1) (human) КЗ ҰВ-Amyloid (1-42) өДҹo»оРФРОКҪЎЈҝЙУГЧчкҺРФҢҰХХЎЈ |
| ҫЮЧФКЙПакP®aЖ· |
| ЧФКЙјӨ»о„© °ьә¬ RapamycinЈ¬MG-132 өИ¶а·NУРР§өДЧФКЙјӨ»о„©ЎЈ |
| ЧФКЙТЦЦЖ„© °ьә¬ 3-Methyladenine (3-MA) Ј¬Resatorvid (TAK-242) өИҪь 200 ·NУРР§өДЧФКЙТЦЦЖ„©ЎЈ |
| ЧФКЙ»ҜәПОпҺм КХдӣБЛ 900+ ·NЧФКЙРЕМ–НЁВ·ПакPөД®aЖ·Ј¬КЗСРҫҝЧФКЙПакPХ{ҝШј°јІІЎөДУРУГ№ӨҫЯЎЈ |
…ўҝјОД«I
1. Evripidis Gavathiotis, Ana Maria Cuerv, et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell2021 May 13;184(10):2696-2714.e25.
2. Susmita Kaushik, Ana Maria Cuervo, et al. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):365-381.
3. Esteban-Martı´nez, L., Sierra-Filardi, E., McGreal,R.S., Salazar-Roa, M, et al. Programmed mitophagy is essential for theglycolytic switch during cell differentiation.EMBO J. 36, 1688-1706.

- PARPТЦЦЖ„©OlaparibөДЧчУГҷCАнј°ФЪҝЖСРЦРөД‘ӘУГ
- БЧЦ¬ҫЫТТ¶юҙјёКВ¶МЗ(DSPE-PEG2000-Mannose)өДҪYҳӢЎўМШРФЕc‘ӘУГ
- ТЯГзЧф„©QS-21өДЧчУГҷCАнј°ФЪТЯГзСРҫҝЦРөД‘ӘУГ
- ЙъОпЦёКҫ„©Чо¶МЕарB•rйgЈЁMITЈ©ңy¶ЁИ«РВ·Ҫ°ёҪйҪB
- ReadiLinҝ№уwҳЛУӣФҮ„©әРөДғһ„Эј°‘ӘУГҲцҫ°
- MST јјРgЦъБҰлyјғ»Ҝө°°ЧөДУHәНБҰ·ЦОц
- ТЦЦЖ„©BIIB021ФЪјҡ°ыҢҚтһәН„УОпҢҚтһөДИЬҪв·Ҫ°ё
- жңГ№УHәНЛШЕјВ“ОпөДғһ„Э
- ОчГАҪЬіЙ№Ұ…ўјУөЪ¶юК®ЛДҢГЦРҮшЙъОпЦЖЖ·ҙу•ю
- 2025AACRДк•юҲAқMВдД»Ј¬MCE”yФҮ„©®aЖ·ББПаЦҘјУёз
- ОчГАҪЬіЙ№Ұ…ўјУөЪК®ҢГЙъОпЦЖЛҺ·Җ¶ЁРФХ“үҜ
- Н¶ёеУР¶Y-2025 MCEЦРҮшЙъГьҝЖҢWСРҫҝҙЩЯMӘ„ХэКҪҶў„У
- °¬ӮҘНШ&°ІНШЛј•юЧhСыХҲЈәјҜІЙ•rҙъөДҸНлsЦЖ„©РВ·ҪПт
- й_ҢWјҫЈ¬ҝЖСРёЈАыөҪ!ұҫЙъЙъОпМбИЎФҮ„©әРПЮ•rМШ»Э
- Оч°І°ЩОһЙъОпАТ°·ИҫБППЮ•rҙЩдNЈ¬ЩҸЩIјҙГвЩMЛНлҠУ°Жұ
- ГАөВВ•НЖіцНЁЯ^LC-MS/MS”UҙуРВЙъғәәYІйөДҷzңyФҮ„©


Copyright(C) 1998-2025 ЙъОпЖчІДҫW лҠФ’Јә021-64166852;13621656896 E-mailЈәinfo@bio-equip.com